Introduction
The prognosis of childhood cancer has improved
significantly, and the number of long-term survivors has increased
in recent years, mainly due to improvements in multidisciplinary
therapy, including neoadjuvant chemotherapy, radiation therapy and
surgical techniques. With the increase in the number of long-term
survivors, the development of latent treatment-related adverse
effects, such as secondary cancer, has generated new problems.
Secondary cancers are defined as histologically
distinct malignancies that develop at least 2 months after the
completion of treatment for primary cancer. Secondary cancers occur
due to genetic factors, as well as previous chemotherapy and/or
radiation therapy.
We herein present three cases of secondary
osteosarcoma that developed after treatment for childhood cancer in
our hospital (Tables I and II) and report on their characteristic
clinical features, diagnosis, treatment and outcomes.
 | Table I.Primary cancer. |
Table I.
Primary cancer.
| Case | Age, years | Histology | Site | Chemotherapy,
cumulative doses | Radiation |
|---|
| 1 | 4 | Medulloblastoma | Cerebellar
vermis | VCR, 0.6
mg/m2, CPM, 12,000 mg/m2 | Yes |
|
|
|
|
| CDDP, 360
mg/m2, VP16, 14,000 mg/m2 | 32 Gy (brain) |
|
|
|
|
| TEPA, 800
mg/m2, LPAM, 280 mg/m2, | 18 Gy (spine) |
|
|
|
|
| MTX (intrathecal
injection), 72 mg/body |
|
| 2 | 9 | Germ cell tumor | Anterior
mediastinum | IFM, 18
g/m2, CBDCA, 4,500 mg/m2 | None |
|
|
|
|
| CDDP, 400
mg/m2, VP-16, 5,100 mg/m2 |
|
|
|
|
|
| BLM, 45
mg/m2, PTX, 650 mg/m2 |
|
| 3 | 1 | Anaplastic
astrocytoma | Medulla
oblongata | VCR, CPM, CDDP,
VP-16, TEPA, L-PAM | Yes |
|
|
|
|
| (Cumulative doses
are not applicable) | 40 Gy (brain) |
| Table II.Secondary cancer. |
Table II.
Secondary cancer.
| Case | Interval | Histology | Site | Prognosis
follow-up |
|---|
| 1 | 6 years | Chondroblastic
osteosarcoma | Proximal left
femur | CDF 6 years |
| 2 | 5 years | Osteoblastic
osteosarcoma | Distal right
femur | CDF 2 years |
| 3 | 8 years | Osteoblastic
osteosarcoma | Proximal right
tibia | DOD 6 months |
The patients and their parents provided their
consent for the publication of patient data.
Case reports
Case 1
An 11-year-old male patient experienced pain in the
left thigh for >2 months. The family history provided no
indication of a genetic predisposition to bone tumors. When the
patient was 4 years old, he developed medulloblastoma in the
cerebellar vermis and was treated by tumor resection and,
postoperatively, whole-brain irradiation (18 Gy), localized
irradiation (32 Gy) and high-dose intensive chemotherapy. The
patient was administered four cycles of chemotherapy with
vincristine, cyclophosphamide, cisplatin and etoposide, and one
cycle with thiotepa and melphalan. The patient also received six
intrathecal injections of dexamethasone and methotrexate.
Furthermore, etoposide was administered for 2 years due to
intrathecal metastasis. The patient remained disease-free for 6
years and 3 months after the last cycle of chemotherapy.
Six years after treatment for medulloblastoma, the
patient experienced gradually increasing pain in the left thigh. At
initial hospital presentation, muscle pain was suspected, as a
plain radiograph revealed no abnormalities. However, the pain
persisted. The patient visited another hospital 2 months after
symptom onset, and a bone tumor of the left femur was identified on
X-ray imaging. At that point, the patient was referred to Osaka
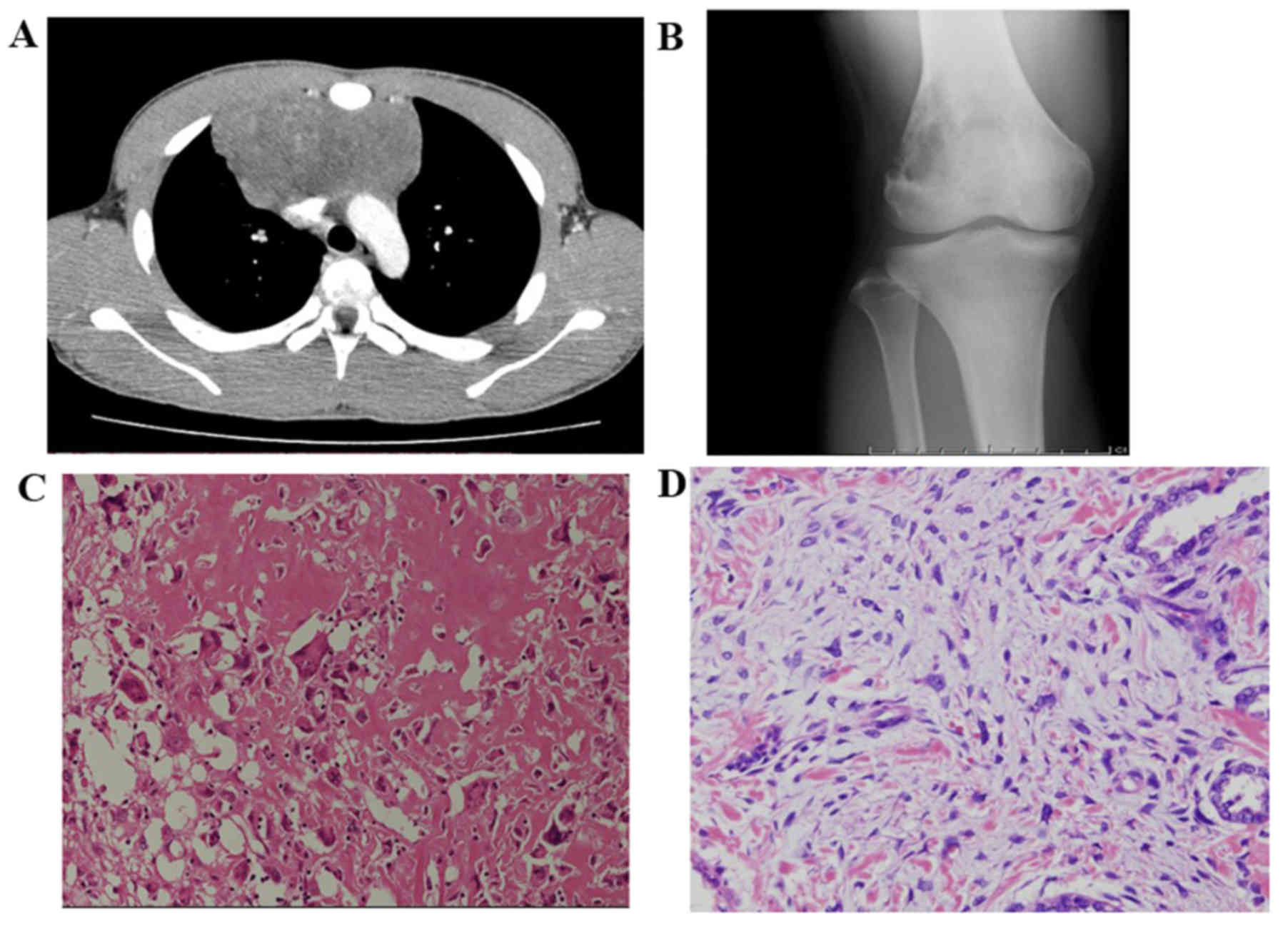
City General Hospital (Osaka, Japan) on December 2008. X-ray
imaging revealed a lytic lesion with spiculated periosteal reaction
in the left proximal femoral shaft (Fig.
1A). Coronal T1-weighted and T2-weighted magnetic resonance
(MR) images revealed a heterogeneously low-intense (T1) and
heterogeneously hyperintense (T2) mass extending from the proximal
femoral bone marrow to the adjacent soft tissues (Fig. 1B). Pathological diagnosis of the
biopsy specimen from the extraosseous lesions revealed
chondroblastic osteosarcoma, with spindle cells in the cartilage
lacunae and a cartilage-like matrix (Fig. 1C). One cycle of chemotherapy with
adriamycin (60 mg/m2) and cisplatin (100
mg/m2), and another with ifosfamide (10 g/m2)
were administered. However, the tumor progressed due to treatment
resistance. Consequently, a wide resection of the bone tumor and
reconstruction with artificial implants were performed. The
definitive diagnosis was chondroblastic osteosarcoma, and the
secondary cancer was suspected to be associated with chemotherapy,
as the histopathological pattern was different from that of the
primary cancer (Fig. 1D).
Postoperatively, two cycles of chemotherapy with methotrexate (132
g/m2), pirarubicin (40 mg/m2), carboplatin
(400 mg/m2), irinotecan (100 mg/m2) and
ifosfamide (10 g/m2) were administered. At the last
follow-up, on February 2015, the patient had remained disease-free
for 6 years.
Case 2
A 14-year-old male patient experienced pain in the
right knee for >3 months. The family history provided no
indication of a genetic predisposition to bone tumors. When the
patient was aged 9 years, he developed a germ cell tumor in the
anterior mediastinum (Fig. 2A).
Preoperatively, the patient was treated with high-dose intensive
chemotherapy including three cycles of bleomycin, etoposide and
cisplatin, one cycle of paclitaxel, ifosfamide and cisplatin, and
one cycle of etoposide and carboplatin (Table I). Postoperatively, two cycles of
chemotherapy with paclitaxel and ifosfamide, and two cycles with
etoposide and carboplatin were administered. The patient remained
disease-free for 4 years and 6 months after the last cycle of
chemotherapy.
Five years after receiving treatment for the germ
cell tumor, the patient experienced gradually increasing pain in
the right knee. The presence of a bone tumor was confirmed at a
local hospital, followed by immediate referral to Osaka City
General Hospital (Osaka, Japan) in February 2013. X-ray imaging
revealed a lytic lesion in the distal right femur (Fig. 2B). MR scanning was not performed due
to the introduction of a stainless-steel wire in the sternum during
the previous surgery. Computed tomography (CT) revealed the
presence of an osteolytic lesion in the distal femoral metaphysis.
Pathological diagnosis of the biopsy specimen from the lesion
confirmed malignancy, suspected to be a sarcomatoid carcinoma.
Preoperatively, the patient received two cycles of chemotherapy. He
then underwent wide resection of the tumor and reconstruction with
artificial knee joint replacement. The definitive diagnosis was
osteoblastic osteosarcoma (Fig. 2C),
and secondary cancer associated with chemotherapy was suspected, as
the histopathological pattern was different from that of the
primary cancer (Fig. 2D).
Postoperatively, four cycles of chemotherapy were administered with
240 mg/m2 cyclophosphamide, 36 g/m2
ifosfamide, 450 mg/m2 pirarubicin and 132
g/m2 methotrexate pre- and postoperatively, according to
CCG-7921 and POG-9351 (1). At the
last follow-up, on March 2015, the patient had remained
disease-free for 2 years.
Case 3
A 9-year-old male patient experienced pain in the
right knee for >1 month. The family history provided no
indication of a genetic predisposition to bone tumors. At the age
of 1 year, the patient developed anaplastic astrocytoma in the
medulla oblongata and was treated with localized brain irradiation
(40 Gy) and high-dose intensive chemotherapy. Four cycles of
chemotherapy with vincristine, cyclophosphamide, cisplatin,
thiotepa and melphalan, and 14 cycles of etoposide were
administered (Table I). The patient
remained disease-free for 6 years after the last cycle of
chemotherapy.
Eight years after treatment for the primary cancer,
the patient experienced gradually increasing pain in the right
knee. At first, sciatica was suspected at a local hospital.
Sagittal T1-weighted and sagittal T2-weighted MR images revealed a
heterogeneously hyperintense (T1) and isointense (T2) mass in the
intradural region at the L3 level (Fig.
3A). Astrocytoma recurrence was suspected, and the patient was
referred to Osaka City General Hospital (Osaka, Japan) in March
2010. X-ray imaging revealed an ossifying lesion with periosteal
reaction in the proximal right tibia. Coronal T1-weighted and
T2-weighted MR images revealed a heterogeneously hypointense (T1)
and heterogeneously hyperintense (T2) mass extending from the
proximal tibial metadiaphysis to the adjacent soft tissues
(Fig. 3B). The patient underwent
intradural tumor resection and anaplastic astrocytoma was
pathologically diagnosed (Fig. 3C);
however, the pathological diagnosis of the biopsy specimen from the
right proximal tibial lesion was osteoblastic osteosarcoma
(Fig. 3D). High-dose intensive
chemotherapy, including ifosfamide (6 g/m2), pirarubicin
(40 mg/m2) and cisplatin (100 mg/m2), was
administered. However, due to rapid tumor growth, thigh amputation
was performed ~2 months after admission. Despite subsequent
multiple regimens of aggressive chemotherapy including pirarubicin
and cisplatin, ifosfamide and etposide, and etposide and
temozolomide, the tumor progressed quickly. The patient eventually
succumbed to disseminated metastatic disease, including the
worsening of lung metastasis in September 2010.
Discussion
We herein report three cases of secondary
osteosarcoma that developed after treatment for a childhood cancer.
Of all the primary malignant bone tumors, osteosarcoma is the most
common, accounting for 19.2% of all primary malignant bone tumors.
However, its absolute incidence is low, at 5.6 cases per million
population annually (2–4).
While osteosarcoma may occur as a secondary
malignancy during childhood, its incidence is very low. In the
large-scale Childhood Cancer survey, osteosarcoma as a secondary
malignancy was reported in 31 of 14,372 survivors of childhood
cancer (5). The incidence of
osteosarcoma as a secondary malignancy following childhood cancer
is expected to rise accordingly, due to the increase in the number
of childhood cancer survivors. While primary osteosarcoma treatment
is based on a clearly defined protocol including chemotherapy and
surgery, the treatment protocol for osteosarcoma as a secondary
neoplasm is not well-established. The 5-year survival rate
associated with conventional osteosarcoma is reportedly 60–70%
(6,7). The prognosis of patients with secondary
bone sarcomas is generally poor. However, Bielack et al
(8) reported that the prognosis of
secondary osteosarcoma may increase to match that of the otherwise
comparable primary osteosarcoma. In that study, chemotherapy was
administered according to the contemporary Cooperative Osteosarcoma
Study Group protocols for secondary osteosarcoma (9), including surgery and multiagent
chemotherapy. Furthermore, Yonemoto et al (10) reported that 7 of 9 patients (77.8%)
with osteosarcoma as a secondary cancer survived without disease
(mean follow-up period, 10.9 years); therefore, the prognosis of
osteosarcoma occurring as a secondary malignancy following
treatment for childhood cancer may be more favorable compared with
that of conventional osteosarcoma.
Secondary cancer is defined as a histologically
distinct malignancy, developing at least 2 months after the
completion of treatment for the primary cancer (11). The cumulative incidence of secondary
cancer is 15% at 20 years after the diagnosis of the primary
cancer, and the incidence risk remains increased for >20 years
(12). This represents a 10-fold
increased risk of cancer among cancer survivors, compared with the
general population (13). In
addition, the risk of secondary bone tumors has been reported to be
133 times higher compared with that of the general population, with
an estimated 20-year cumulative risk of 2.8% (14).
The prognosis of childhood cancer has markedly
improved since the 1970s, mainly due to improvements in
multidisciplinary therapy, including neoadjuvant chemotherapy,
radiation therapy and surgical techniques. However, with the
increase in the number of long-term survivors, the prevalence of
latent treatment-related adverse effects, such as secondary
malignancy, has increased (15).
Medulloblastoma (case 1) accounts for ~10% of all
childhood brain tumors. These tumors occur exclusively in the
posterior fossa and have a propensity for leptomeningeal spread.
Treatment includes a combination of surgery and radiation therapy
(patients aged >3 years). Adjuvant chemotherapy is recommended
for all patients. Patients aged >3 years are stratified based on
the volume of postoperative metastases (reference value: 1.5
cm2) or absence of metastases into standard- and
high-risk categories, with long-term survival rates of ~85 and 70%,
respectively. Outcomes are inferior in infants and children aged
<3 years (16). The patient in
case 1 was 4 years old and the primary tumor was resected
completely, but intrathecal metastasis was present. Thus, he was
deemed a high-risk patient. The radiation dose was reduced to 18
Gy, and an alkylator-based, dose-intensive chemotherapy regimen was
administered according to the Children's Oncology Group study in
young children (aged 3–7 years) (16,17).
Mediastinal germ cell tumors (case 2) are a rare
group of germ cell tumors that account for <5% of all germ cell
malignancies. Histologically, they may be divided into two
categories, namely seminomas and non-seminomas (18). The long-term cure rate of patients
with histologically pure seminomas in the mediastinum is almost
90%; only 45% of patients with mediastinal non-seminoma survive for
5 years, and their outcomes are worse compared with those of
patients with seminomas in the gonads or retroperitoneal primary
lesions (19). The pathological
diagnosis of the primary cancer of the patient in case 2 was
non-seminoma. The treatment included conventional chemotherapy with
the bleomycin + etoposide + cisplatin and paclitaxel + ifosfamide +
cisplatin protocols, with modifications.
Overall, the age-adjusted incidence of anaplastic
astrocytoma is 3.5 per million persons/year. The incidence of
anaplastic astrocytoma (case 3) increases with age, and in children
aged 0–15 years, the rate is 0.9 per million persons/year. The
corresponding rates are 2.6 in young adults (16–39 years) and 4.7
in older adults (40–64 years). The overall age-standardized 5- and
10-year relative survival rates of populations with anaplastic
astrocytoma are 23.6 and 15.1%, respectively, while the respective
5- and 10-year survival rates in children are 32.1 and 24.6%. The
effect of age on survival is present for only the first 3 years
post-diagnosis, after which time age no longer affects
cancer-specific survival rates (20). The patient in case 3 received
localized irradiation and a high-dose intensive chemotherapy
protocol that included cyclophosphamide, vincristine, etoposide and
cisplatin, put forth by the Pediatric Oncology Group for the
treatment of children aged <3 years with malignant astrocytoma,
with modifications (21).
At least two factors, namely genetic factors and
acquired conditions related to treatment modalities, must be
considered as the possible causes of secondary malignancy.
Genetic factors include the presence of Li-Fraumeni
syndrome (LFS) and retinoblastoma (22,23).
Previous studies have demonstrated that childhood cancer survivors
with a family history of cancer, particularly the presence of LFS,
carry an increased risk of developing secondary cancer (24,25). In
all three patients in the present report, the familial history did
not fit the clinical diagnostic criteria of LFS or retinoblastoma
(22,23).
As regards acquired factors, radiation and
chemotherapy have been implicated in the past. Therapy-related
solid secondary malignancy is strongly associated with radiation.
The risk of solid secondary malignancy is highest when the exposure
to radiation occurs at a younger age, and it increases with the
total dose of radiation (13,14).
Some well-established radiation-related solid secondary
malignancies include breast, lung and thyroid cancer, brain tumors,
soft tissue sarcomas, and malignant bone tumors, such as Ewing's
sarcoma (26). Our patients had not
received previous radiation therapy to the anatomical site of the
secondary malignancy.
It has been suggested that chemotherapy may cause
secondary malignancy in young cancer survivors (26,27).
Bhatia et al (26) concluded
that the use of alkylating agents and topoisomerase II inhibitors
for childhood cancer increases the subsequent risk of secondary
tumors. Hawkins et al (28)
reported that the risk of alkylating agent-related secondary
malignancy is dose-dependent.
Mutagenicity is associated with the ability of
alkylating agents to form crosslinks and/or transfer alkyl groups
to form DNA monoadducts (29).
Topoisomerase II catalyzes the relaxation of supercoiled DNA by
covalently binding and transiently cleaving and re-ligating both
strands of the DNA helix. DNA topoisomerase II inhibitors stabilize
the enzyme-DNA covalent intermediate, decrease the relegation rate,
and cause chromosomal breakage (30). In our three cases, alkylating agents
(ifosfamide and cyclophosphamide) and a topoisomerase II inhibitor
(etoposide) had been administered for the primary cancer. While it
is impossible to definitively establish a link between previous
chemotherapy and secondary malignancies, an association may well
exist.
The interval from the diagnosis of primary
malignancy to the development of secondary malignancy may be 8–10
years (27). In our patients, a
period of ~7 years elapsed between the initial diagnosis of primary
cancer and the detection of a possible secondary malignancy. Thus,
long-term follow-up may be necessary to identify the development of
a secondary malignancy in young survivors of malignant tumors in
order to monitor latent treatment-related adverse effects.
In conclusion, we herein presented three cases of
osteosarcoma developing several years after the primary
malignancies. Medical professionals, including orthopedic
oncologists, as well as patients, should be aware of the
possibility of the occurrence of a secondary malignancy as a
potential adverse latent effect. Long-term follow-up is crucial for
young survivors, even after successful treatment of the primary
cancer.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
Conception or design of the work: AS; data
collection: AS, NO, TI, NT, JH, CN; data analysis and
interpretation: AS, CN; drafting the article: AS; critical revision
of the article: MA, MH; final approval of the version to be
published: HN.
Ethics approval and consent to
participate
Study approval was obtained from the institution,
and all investigations were conducted in accordance with the
ethical principles of research.
Patient consent for publication
The patients and their families provided consent for
the publication of patient data.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MR
|
magnetic resonance
|
|
CT
|
computed tomography
|
References
|
1
|
Meyers PA, Schwartz CL, Krailo M,
Kleinerman ES, Betcher D, Bernstein ML, Conrad E, Ferguson W,
Gebhardt M, Goorin AM, et al: Osteosarcoma: A randomized,
prospective trial of the addition of ifosfamide and/or muramyl
tripeptide to cisplatin, doxorubicin and high-dose methotrexate. J
Clin Oncol. 23:2004–2011. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Raymond AK, Ayala AG and Nuutila S:
Conventional osteosarcomaWorld Health Organization classification
of tumors: Pathology and genetics; tumors of soft tissue and bone.
Fletcher CDM, Unni KK and Mertens F: International Agency for
Research on Cancer; Lyon: pp. 24–70. 2002
|
|
3
|
Uni KK and Inwards CY:
OsteosarcomaDahlin's bone tumors. 6th edition. Uni KK and Inwards
CY: Wolters Kluwer, Lippincott Williams and Wilkins; Philadelphia:
pp. 122–157. 2010
|
|
4
|
Arndt CA and Crist WM: Common
musculoskeletal tumors of childhood and adolescence. N Engl J Med.
341:342–352. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Henderson TO, Whitton J, Stovall M,
Mertens AC, Mitby P, Friedman D, Strong LC, Hammond S, Neglia JP,
Meadows AT, et al: Secondary sarcomas in childhood cancer
survivors: A report from the childhood cancer survivor study. J
Natl Cancer Inst. 99:300–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smith MA, Seibel NL, Altekruse SF, Ries
LA, Melbert DL, O'Leary M, Smith FO and Reaman GH: Outcomes for
children and adolescents with cancer: Challenges for the
twenty-first century. J Clin Oncol. 28:2625–2634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cho Y, Jung GH, Chung SH, Kim JY, Choi Y
and Kim JD: Long-term survivals of stage IIb osteosarcoma: A
20-year experience in a single institution. Clin Orthop Surg.
3:48–54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bielack SS, Kempf-Bielack B, Heise U,
Schwenzer D and Winkler K: Combined modality treatment for
osteosarcoma occurring as a second malignant disease. Cooperative
German-Austrian-Swiss Osteosarcoma Study Group. J Clin Oncol.
17:11641999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bielack S, Beck J, Delling G, Gerein V,
Grümayer R, Hiddemann W, Jobke A, Jürgens H, Kornhuber G, Kotz R,
et al: Neoadjuvant chemotherapy of osteosarcoma. Results of the
cooperative studies COSS-80 and COSS-82 after 7 and 5 years. Klin
Padiatr. 201:275–284. 1989.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yonemoto T, Hosono A, Iwata S, Kamoda H,
Hagiwara Y, Fujiwara T, Kawai A and Ishii T: The prognosis of
osteosarcoma occurring as second malignancy of childhood cancers
may be favorable: Experience of two cancer centers in Japan. Int J
Clin Oncol. 20:613–616. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pizzo PA and Poplack DG: Principles and
practice of pediatric oncology. Wolters Kluwer; Sixth edition.
2015
|
|
12
|
Meadows AT, Friedman DL, Neglia JP,
Mertens AC, Donaldson SS, Stovall M, Hammond S, Yasui Y and Inskip
PD: Second neoplasm in survivors of childhood cancer: Findings from
the childhood cancer survivor study cohort. J Clin Oncol.
27:2356–2362. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Neglia JP, Friedman DL, Yasui Y, Mertens
AC, Hammond S, Stovall M, Donaldson SS, Meadows AT and Robison LL:
Second malignant neoplasms in five-year survivors of childhood
cancer: Childhood cancer survivor study. J Natl Cancer Inst.
93:618–629. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tucker MA, D'Angio GJ, Boice JD Jr, Strong
LC, Li FP, Stovall M, Stone BJ, Green DM, Lombardi F, Newton W, et
al: Bone sarcomas linked to radiotherapy and chemotherapy in
children. N Engl J Med. 317:588–593. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hudson MM: Late complications after
leukemia therapy. Cambridge University Press; pp. 701–713. 2012
|
|
16
|
Millard NE and De Braganca KC:
Medulloblastoma. J Child Neurol. 31:1341–1353. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gajjar A, Chintagumpala M, Ashley D,
Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin
MJ, et al: Risk-adapted craniospinal radiotherapy followed by
high-dose chemotherapy and stem-cell rescue in children with newly
diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term
results from a prospective, multicentral trial. Lancet Oncol.
7:813–820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chetaille B, Massard G and Falcoz PE:
Mediastinal germ cell tumors. Anatomopathology, classification,
teratomas and malignant tumors. Rev Pneumol Clin. 66:63–70.
2010.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bokemeyer C, Nichols CR, Droz JP, Schmoll
HJ, Horwich A, Gerl A, Fossa SD, Beyer J, Pont J, Kanz L, et al:
Extragonadal germ cell tumors of the mediastinum and
retroperitoneum: Results from an international analysis. J Clin
Oncol. 20:1864–1873. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smoll NR and Hamilton B: Incidence and
relative survival of anaplastic astrocytomas. Neuro Oncol.
16:1400–1407. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duffner PK, Horowitz ME, Krischer JP,
Friedman HS, Burger PC, Cohen ME, Sanford RA, Mulhern RK, James HE,
Freeman CR, et al: Postoperative chemotherapy and delayed radiation
in children less than three years of age with malignant brain
tumors. N Engl J Med. 328:1725–1731. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li FP, Fraumeni JF Jr, Mulvihill JJ,
Blattner WA, Dreyfus MG, Tucker MA and Miller RW: A cancer family
syndrome in twenty-four kindreds. Cancer Res. 48:5358–5362.
1988.PubMed/NCBI
|
|
23
|
Kay RM, Eckardt JJ and Mirra JM:
Osteosarcoma and Ewing's sarcoma in a retinoblastoma patient. Clin
Orthop Relat Res. 323:284–287. 1996. View Article : Google Scholar
|
|
24
|
Andersson A, Enblad G, Tavelin B,
Björkholm M, Linderoth J, Lagerlöf I, Merup M, Sender M and Malmer
B: Family history of cancer as a risk factor for second
malignancies after Hodgkin's lymphoma. Br J Cancer. 98:1001–1005.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hisada M, Garber JE, Fung CY, Fraumeni JF
Jr and Li FP: Multiple primary cancers in families with Li-Fraumeni
syndrome. J Natl Cancer Inst. 90:606–611. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bhatia S and Sklar C: Second cancers in
survivors of childhood cancer. Nat Rev Cancer. 2:124–132. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Longhi A, Ferrari S, Tamburini A, Luksch
R, Fagioli F, Bacci G and Ferrari C: Late effects of chemotherapy
and radiotherapy in osteosarcoma and Ewing sarcoma patients: The
Italian Sarcoma Group Experience (1983–2006). Cancer.
118:5050–5059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hawkins MM, Wilson LM, Burton HS, Potok
MH, Winter DL, Marsden HB and Stovall MA: Radiotherapy, alkylating
agents, and risk of bone cancer after childhood cancer. J Natl
Cancer Inst. 88:270–278. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thirman MJ and Larson RA: Therapy-related
myeloid leukemia. Hematol Oncol Clin North Am. 10:293–320. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Corbett AH and Osheroff N: When good
enzymes go bad: Conversion of topoisomerase II to a cellular toxin
by antineoplastic drugs. Chem Res Toxicol. 6:585–597. 1993.
View Article : Google Scholar : PubMed/NCBI
|