Introduction
Immune thrombocytopenic purpura (ITP) is
characterized by antibody- and immune-precipitated platelet
destruction, occasionally causing life-threatening hemorrhage. ITP
is a frequent complication of hematological malignancies,
particularly chronic lymphoid leukemia (1); however, it is less common in
non-Hodgkin lymphoma (NHL), affecting only 0.76% of the patients
(2). Thus, the etiology and
treatment of NHL-associated ITP have not been clearly determined,
although this is a potentially life-threatening condition that is
difficult to manage.
Only 10 cases of diffuse large B-cell lymphoma
(DLBCL), which is the most common type of NHL, complicated by ITP
have been reported to date (2).
T-cell/histiocyte-rich B-cell lymphoma (T/HRBCL) is an uncommon
variant of DLBCL characterized by a limited number of atypical B
cells in a background of abundant T cells and histiocytes (3,4). A case
of T/HRBCL complicated by symptoms mimicking autoimmune disease was
recently reported (5). We herein
describe what is, to the best of our knowledge, the first case of
T/HRBCL-associated ITP successfully treated by chemotherapy.
Case report
A 54-year-old man presented to Kobe City Medical
Center General Hospital (Kobe, Japan) in May 2017 with a fever of
38.9°C, bilateral cervical and right axillary lymphadenopathies,
petechial purpura on the upper and lower limbs and intraoral
mucosal hemorrhage. The patient reported a history of fever and
lymphadenopathy that started 1 month prior to hospitalization, and
he was admitted to our hospital for thorough examination and
treatment. The patient had no symptoms indicative of collagen
disease, such as skin rash or joint pain. His liver and spleen were
not palpable. The laboratory data on admission are summarized in
Table I. Briefly, complete blood
count and biochemical tests revealed the following: Hemoglobin,
12.2 g/dl; platelet count, 0.5×109/l; reticulated
platelets, 59%; white blood cell count, 10.1×109/l;
lactate dehydrogenase (LDH), 315 U/l; soluble interleukin-2
receptor, 25,045 U/ml; platelet-associated immunoglobulin G, 462.2
ng/107 cells; antinuclear antibody, negative; and
Epstein-Barr virus antibody and human immunodeficiency virus,
negative. Computed tomography examination revealed bilateral
cervical (25 mm), bilateral axillary (15 mm), mediastinal (19 mm)
and para-aortic (20 mm) lymphadenopathies (Fig. 1). A bone marrow biopsy revealed mild
hypercellularity, with an increase in the number of megakaryocytes;
however, pathological lymphocytes or phagocytes were not
identified. Flow cytometric analysis and molecular analysis
confirmed the absence of clonal malignant cell invasion. The
suspected diagnosis was immune thrombocytopenic purpura (ITP),
based on the thrombocytopenia, normal white blood cell count, mild
anemia, increase in reticulated platelets, and lack of evidence of
malignant cell invasion, hemophagocytic syndrome, or other cause of
thrombocytopenia. Intravenous immunoglobulins were administered,
but did not improve the platelet count. A biopsy of a right
axillary lymph node was performed and the lymph node structure was
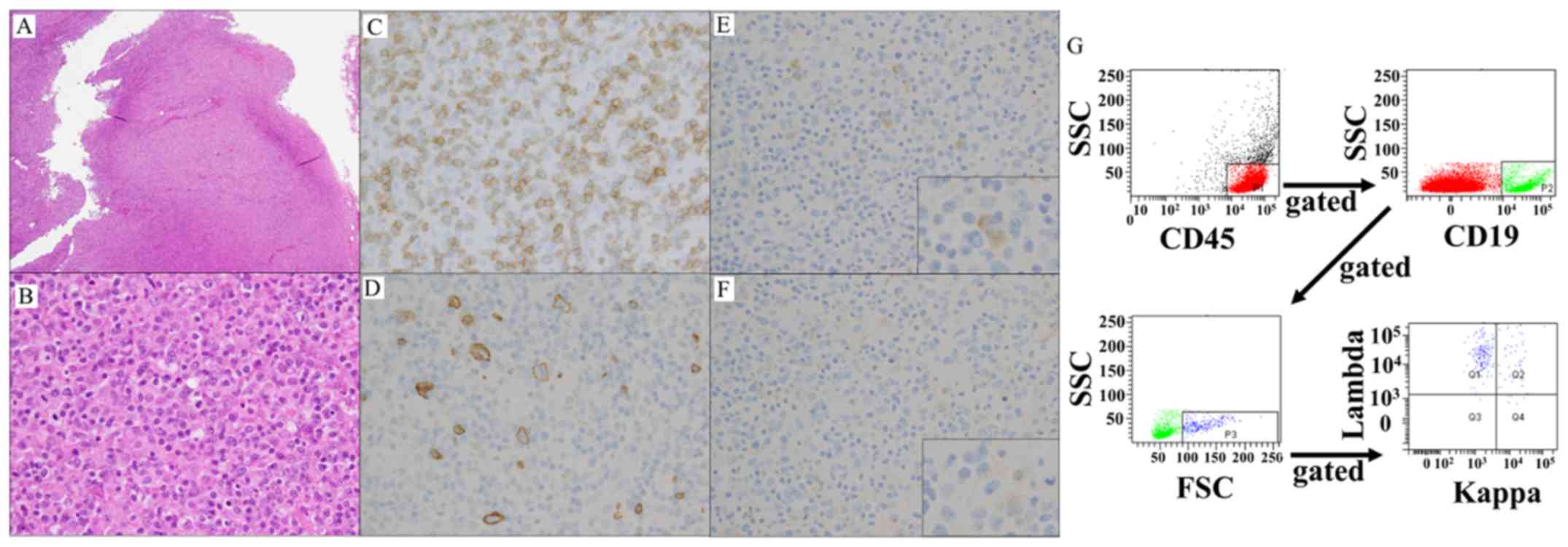
found to be almost completely effaced by a diffuse infiltrate
(Fig. 2A). A small number of
scattered large atypical cells were found among numerous mature
lymphoid cells (Fig. 2B).
Immunohistochemical examination revealed expression of CD20 on the
large malignant cells and CD3 on the small cells of the background.
CD30, CD15 and Epstein-Barr virus early small ribonucleic acids
were absent in both the large and small lymphocytes (Fig. 2C and D). The infiltrating large cells
were positive for λ but negative for κ light chain (Fig. 2E-G). Immunoglobulin heavy chain
rearrangement was not detected, and the karyotype of the lymphoma
cells was 46,XY. Therefore, the patient was diagnosed with T/HRBCL
of Ann Arbor stage IIIB and an International Prognostic Index of 2
points (stage and LDH).
 | Table I.Patient's laboratory data on
admission. |
Table I.
Patient's laboratory data on
admission.
| Hematology | Patient data | Normal range |
|---|
| White blood cell
count (/µl) | 10,100 | 3,900–9,800 |
|
Neutrophils (%) | 54.0 |
|
|
Eosinophils (%) | 0.5 |
|
| Basophils
(%) | 0.0 |
|
| Monophils
(%) | 11.0 |
|
|
Lymphocytes (%) | 33.0 |
|
| Other
(%) | 1.5 |
|
| Red blood
cell count (×104/µl) | 398 | 410–570 |
|
Hemoglobin (g/dl) | 12.2 | 13.4–17.6 |
| Platelet
count (×104/µl) | 0.5 | 13.0–37.0 |
|
Reticulated platelets (%) | 59 | 0–10 |
| Coagulation |
| PT
(%) | 72.1 | 80.0–125.0 |
| APTT
(sec) | 37.1 | 24.3–38.9 |
| D-dimer
(µg/ml) | 17.90 | <1.00 |
|
Fibrinogen (mg/dl) | 149 | 180–320 |
| Tumor marker |
| sIL-2R
(U/ml) | 25,045 | 122–496 |
| Biochemistry |
| Total
protein (g/dl) | 5.4 | 6.5–8.5 |
| Albumin
(g/dl) | 3.1 | 3.9–4.9 |
| Total
bilirubin (mg/dl) | 1.2 | 0.2–1.2 |
| AST
(U/l) | 55 | 8–40 |
| ALT
(U/l) | 29 | 8–40 |
| LDH
(U/l) | 315 | 120–250 |
| BUN
(mg/dl) | 15.3 | 8.0–20.0 |
|
Creatinine (mg/dl) | 0.66 | 0.60–1.10 |
| Na
(mEq/l) | 135 | 136–148 |
| K
(mEq/l) | 4.0 | 3.5–5.3 |
| Ca
(mg/dl) | 8.3 | 8.0–10.0 |
| CRP
(mg/dl) | 0.16 | 0.00–0.50 |
| Immunological |
| PA-IgG
(ng/107 cells) | 462.2 | <30.2 |
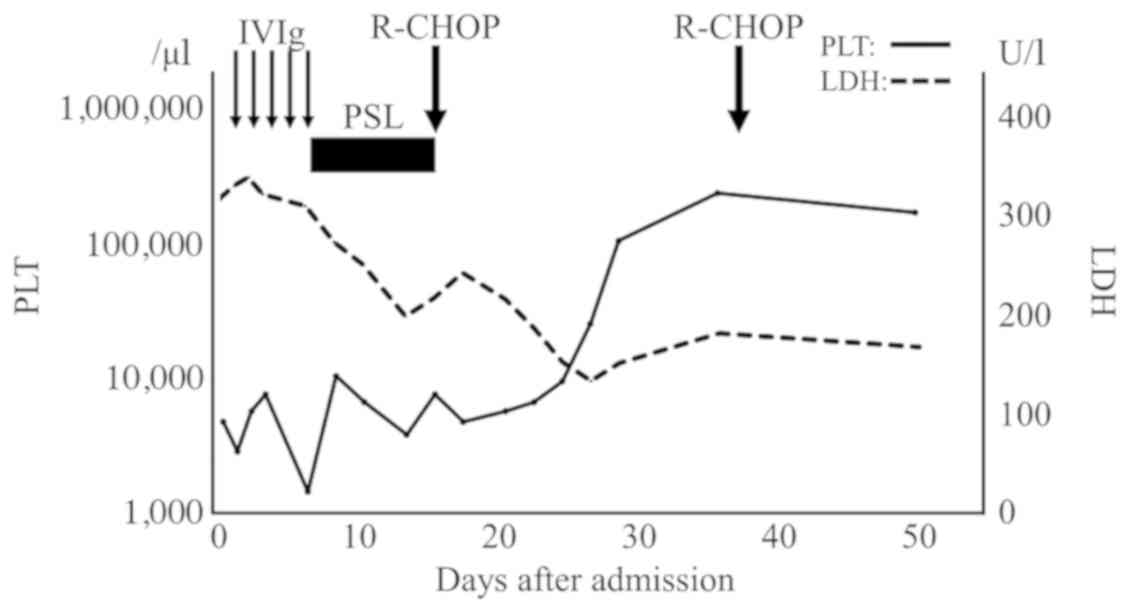
The patient received prednisolone (100 mg) from the
day of biopsy until confirmation of the pathological diagnosis;
thereafter, he was treated with the R-CHOP regimen, which consisted
of rituximab (375 mg/m2), cyclophosphamide (750
mg/m2), doxorubicin (50 mg/m2) and
vincristine [1.4 mg/m2 (max 2 mg/body)] on day 1, and
prednisolone (100 mg/body) on days 1–5. Prompt reduction of the
size of the superficial lymph nodes was observed, whereas the
platelet count started to increase at 10 days and normalized at 36
days after chemotherapy initiation (Fig.
3). Treatment was terminated after three cycles due to
financial difficulties.
The patient visited our hospital again 10 months
after treatment interruption due to fever, fatigue,
lymphadenopathies and bleeding tendency. The laboratory data were
similar to those at first presentation: Hemoglobin, 10.0 g/dl;
platelet count, 0.1×109/l; white blood cell count,
5.4×109/l; LDH, 264 U/l; ferritin, 615 ng/ml (normal
range, 33.8–369.1); and soluble interleukin-2 receptor, 25,972
U/ml. Computed tomography examination revealed diffuse
lymphadenopathy. Re-biopsy was not performed due to the strong
bleeding tendency and the need for urgent treatment. The patient
received three cycles of R-CHOP and two cycles of rituximab (375
mg/m2) and carboplatin (area under the curve = 5) on day
1, plus ifosphamide (1,700 mg/m2) and etoposide (100
mg/m2) on days 1–3 (R-ICE regimen).
The platelet count was increased and this increase
was sustained after the first chemotherapy, whereas positron
emission tomography after the treatment revealed complete remission
of T/HRBCL. Further treatment was not carried out because of a
treatment refusal.
Discussion
We herein report the first case of
T/HRBCL-associated ITP that substantially improved with
chemotherapy. The patient initially received a standard treatment
for ITP that failed to improve the thrombocytopenia, but he
achieved complete remission of ITP following administration of
R-CHOP chemotherapy, indicating the efficacy of R-CHOP for patients
with T/HRBCL-associated ITP. In addition, the reproducibility at
recurrence suggested an association between T/HRBCL and ITP.
No specific criteria for the diagnosis of ITP have
been established to date, and diagnosing patients is a process of
exclusion. Drugs, infection, autoimmune disease, liver diseases and
bone marrow diseases may cause thrombocytopenia. In the present
case, the patient had no diseases that may have caused the
thrombocytopenia except for T/HRBCL, and there was no bone marrow
invasion or hemophagocytic syndrome that may result in
thrombocytopenia. In addition to these observations at the time of
first presentation, the patient had the same presentation at the
time of recurrence. It is difficult to exclude disseminated
intravascular coagulation, but the clinical presentation of this
patient was different from previous reports in terms of very low
platelet count at presentation, no organ abnormalities and
low-intermediate IPI score (6,7). Thus,
the patient was diagnosed with T/HRBCL-associated ITP, although
there was a possibility of complicated disseminated intravascular
coagulation. This diagnosis was supported by the observed platelet
recovery as the lymphoma remitted.
ITP is often associated with chronic lymphoid
leukemia, but it is less common in patients with malignant
lymphoma1 (1,2), with only 10 cases of DLBCL-associated
ITP reported to date (2). T/HRBCL is
an uncommon variant of DLBCL (3,4), and
patients with T/HRBCL are more likely to develop B symptoms and
respond to R-CHOP therapy, with an overall response rate of 56–63%
(8–11). Although the exact mechanism
underlying the association of thrombocytopenia and malignant
lymphoma is unknown, there are certain possible hypotheses. One
such hypothesis is that anti-platelet antibodies are produced by
lymphoma cells. This is supported by findings of platelet antibody
production by DLBCL cells during ITP (12). In the present case, the level of
platelet-associated immunoglobulin G was decreased after R-CHOP
treatment, which supports this hypothesis. In this case of T/HRBCL
complicated by symptoms mimicking systemic lupus erythematosus,
upregulation of innate immune molecules, including interleukin-6,
interleukin-8 and growth-regulated oncogene, induced multiple
autoimmune reactions that ultimately resulted in symptoms similar
to those of systemic lupus erythematosus (5). Unfortunately the cytokine profiles in
the present case were not evaluated, but T/HRBCL-induced innate
immunity may have been associated with the development of ITP.
The standard first-line treatment for severe ITP
includes corticosteroids and/or intravenous immunoglobulin.
However, such treatment has been previously reported to be
ineffective for DLBCL-associated ITP. Uchiyama et al
reported a case of DLBCL-associated ITP in which standard ITP
therapy was not effective, but the patient's platelet count was
improved after R-CHOP (13). Berrang
et al reported a case of isolated DLBCL complicated by ITP.
The patient was treated with radiotherapy, and his platelet count
also improved (14). These cases
suggest that antitumor therapies, such as chemotherapy and
radiotherapy, are more effective for NHL-ITP. Similarly,
intravenous immunoglobulin administration failed to improve the
thrombocytopenia in our patient, but he responded to R-CHOP therapy
at first presentation and recurrence. Hence, chemotherapy should be
further evaluated and considered as first-line therapy for T/HRBCL-
and DLBCL-associated ITP.
Acknowledgements
The authors would like to thank the medical and
nursing staff at the Kobe City Medical Centre General Hospital.
Funding
No funding was received.
Availability of data and materials
All data analyzed during the present study are
included in this published manuscript.
Authors' contribution
TO, YS and TI contributed to the diagnosis and wrote
the manuscript. DY and YI performed the pathological examination.
All the authors have read and approved the final version of this
manuscript.
Ethics approval and consent to
participate
The requirement for review of this case report was
waived by the Institutional Review Board of our hospital.
Patient consent for publication
Written informed consent was obtained from the
patient regarding the publication of the case details and any
associated images.
Competing interests
The authors declare that they have no competing
financial or non-financial interests to disclose.
Glossary
Abbreviations
Abbreviations:
|
CD
|
cluster of differentiation
|
|
DLBCL
|
diffuse large B-cell lymphoma
|
|
ITP
|
immune thrombocytopenic purpura
|
|
NHL
|
non-Hodgkin lymphoma
|
|
T/HRBCL
|
T-cell/histiocyte-rich B-cell
lymphoma
|
References
|
1
|
Váróczy L, Gergely L, Zeher M, Szegedi G
and Illés A: Malignant lymphoma-associated autoimmune diseases - a
descriptive epidemiological study. Rheumatol Int. 22:233–237. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hauswirth AW, Skrabs C, Schützinger C,
Raderer M, Chott A, Valent P, Lechner K and Jäger U: Autoimmune
thrombocytopenia in non-Hodgkin's lymphomas. Haematologica.
93:447–450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abramson JS: T-cell/histiocyte-rich B-cell
lymphoma: Biology, diagnosis, and management. Oncologist.
11:384–392. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Swerdlow SH, Campo E, Pileri SA, Harris
NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz
AD, et al: The 2016 revision of the World Health Organization
classification of lymphoid neoplasms. Blood. 127:2375–2390. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu B and Cheng Y: Upregulation of innate
immune responses in a T cell/histiocyte-rich large B cell lymphoma
patient with significant autoimmune disorders mimicking systemic
lupus erythematosus. Ann Hematol. 93:353–354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Asakura H, Takahashi H, Tsuji H,
Matsushita T, Ninomiya H, Honda G, Mimuro J, Eguchi Y, Kitajima I
and Sakata Y: Post-marketing surveillance of thrombomodulin alfa, a
novel treatment of disseminated intravascular coagulation - safety
and efficacy in 1,032 patients with hematologic malignancy. Thromb
Res. 133:364–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chi S and Ikezoe T: Disseminated
intravascular coagulation in non-Hodgkin lymphoma. Int J Hematol.
102:413–419. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aki H, Tuzuner N, Ongoren S, Baslar Z,
Soysal T, Ferhanoglu B, Sahinler I, Aydin Y, Ulku B and Aktuglu G:
T-cell-rich B-cell lymphoma: A clinicopathologic study of 21 cases
and comparison with 43 cases of diffuse large B-cell lymphoma. Leuk
Res. 28:229–236. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang J, Sun NCJ, Chen YY and Weiss LM:
T-cell/histiocyte-rich large B-cell lymphoma displays a
heterogeneity similar to diffuse large B-cell lymphoma: A
clinicopathologic, immunohistochemical, and molecular study of 30
cases. Appl Immunohistochem Mol Morphol. 13:109–115. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bouabdallah R, Mounier N, Guettier C,
Molina T, Ribrag V, Thieblemont C, Sonet A, Delmer A, Belhadj K,
Gaulard P, et al: T-cell/histiocyte-rich large B-cell lymphomas and
classical diffuse large B-cell lymphomas have similar outcome after
chemotherapy: A matched-control analysis. J Clin Oncol.
21:1271–1277. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Achten R, Verhoef G, Vanuytsel L and De
Wolf-Peeters C: T-cell/histiocyte-rich large B-cell lymphoma: A
distinct clinicopathologic entity. J Clin Oncol. 20:1269–1277.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tan J, Wei J, Ni X and Zhou J: Immune
thrombocytopenic purpura occurred prior to multiple extranodal
diffuse large B cell lymphoma. Platelets. 22:81–83. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Uchiyama M, Sato K and Ikeda T: Diffuse
large B-cell lymphoma complicated with autoimmune thrombocytopenia.
Intern Med. 50:1215–1218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Berrang T, Holloway C, Hart J, Yee A,
Berry B and Kotb R: Successful treatment of non-Hodgkin lymphoma
associated immune thrombocytopenia with involved field
radiotherapy. Hematol Oncol. 31:218–220. 2013. View Article : Google Scholar : PubMed/NCBI
|