Introduction
Neurofibromatosis type 1 (NF1) is also called von
Recklinghausen disease, which is an autosomal dominant disorder
characterized by café-au-lait spots, axillary and/or inguinal
freckles, lisch nodules, and neurofibromas (1). Patients with NF1 occasionally present
with malignant tumors arising from neurofibromas, which are
referred to as malignant peripheral nerve-sheath tumors (MPNSTs),
and the treatment for such a condition is challenging. The 5-year
overall survival rate was approximately 34-64% in several studies.
The mean age at diagnosis is 34.0 years, and patients with NF1 are
younger than those without NF1 (mean age: 28.7 vs. 39.7 years)
(2,3).
Neurofibromatosis is classified into several
subtypes. One subtype is the mosaic localized NF1 (also called NF5
or segmental NF), which is characterized by the local appearance of
neurofibromatosis (4-6).
Although most MPNSTs occur in patients with NF1, only three
patients presented with mosaic localized NF1 (7,8). The
prevalence rate of mosaic localized NF1 is approximately 0.0018%,
whereas that of NF1 ranges from 0.02 to 0.03% (9,10). Such
a result indicates the rarity of mosaic localized NF1.
Herein, we first report the case of MPNST in an
adolescent patient with mosaic localized NF1. Initially, the
patient underwent unplanned excision due to misdiagnosis with a
benign tumor. The postoperative histologic diagnosis was spindle
cell sarcoma; thus, a wide resection was conducted. The presence of
segmental café-au-lait spots and freckles in the unilateral leg was
helpful in diagnosing MPNST associated with mosaic localized NF1.
Furthermore, NF1 microdeletion was confirmed in the café-au-lait
spot, which was consistent with previous reports. Sparse NF1
expression was also observed in the tumor. Finally, the patient was
diagnosed with MPNST with mosaic localized NF1.
Case report
A 16-year-old man presented with pain and a mass on
the medial side of his right knee for 1.5 years prior to when he
visited his first doctor. The tumor was approximately 3 cm in
diameter. He underwent magnetic resonance imaging (MRI) that
revealed a circumscribed soft tissue tumor located in the
subcutaneous tissue. The tumor had an iso-signal intensity on
T1-weighted sequences and high signal intensity on T2-weighted
sequences (Fig. 1). The doctor
diagnosed the tumor as benign and then performed a marginal
resection. However, postoperative histological diagnosis revealed
spindle cell sarcoma. The patient was then referred to our hospital
for a more specialized treatment. We identified dense freckles and
café-au-lait spots on his right inguinal region to the knee
(Fig. 2). He had no family history
of NF1. The segmental freckles led us to suspect MPNST associated
with mosaic localized NF1. Preoperative MRI before additional wide
resection showed that a residual tumor appeared to exist adjacent
to the patella (Fig. 3A and B). We then confirmed the absence of
metastasis and conducted an additional wide resection where part of
the vastus medialis, the medial patellar retinaculum and joint
capsule, and the patella were resected with the residual tumor.
Soft tissue defect was reconstructed with pedicled anterolateral
thigh musculocutaneous flap. There was no evidence of residual
tumor and surgical margin proved to be negative (R0 resection)
(Fig. 3C). The patient did not
present with local recurrence and distant metastasis 1.5 years
after surgery.
The pathologic review of the primary tumor was
carried out with additional immunostainings at our institute
(Vimentin, S100, SOX10, CD68, Ki67, H3K27me3, and NF1). Hematoxylin
and eosin staining of the marginally removed mass was
well-circumscribed and embedded in the subcutis showed spindle cell
proliferation arranged in interlacing fascicules against a chronic
inflammatory background, which comprised cellular areas alternating
with less cellular areas, accompanied by hemangiopericytoma-like
vessels and a small amount of extracellular myxoid matrix (Fig. 4A). Intratumoral necrosis was not
apparent. The tumor cells had moderate-to-severe nuclear atypia
with mitotic figures (count: 15/10 high-power fields) and focal
pleomorphism (Fig. 4B). Unequivocal
lipoblasts were not identified. An immunohistochemical study showed
reactivity with variable cell populations and intensity to the
following markers: Vimentin (Fig.
4C), desmin, αSMA, S100 (Fig.
4D), MDM2, CDK4, factor XIIIa, CD99, α1-antitrypsin, and a Ki67
labeling index of 20% (Fig. 4E); the
tumor cells were diffusely positive for vimentin in contrast to the
form of scattered positive cells (<5%) for S100. The negative
markers and in situ hybridization (ISH) were as follows:
Cytokeratins (AE1/AE3, CAM5.2, CK7, and CK19), caldesmon, myogenin,
neurofilament, protein melan-A, CD21, CD23, CD30, CD31, CD34, CD68
(Fig. 4F), CD117, BCL2, TLE1, INI1,
SOX10 (Fig. 4G), STAT6, PAX5, ER,
PgR, and Epstein-Barr encoding region ISH. The tumor partially lost
the expression of H3K27me3 (Fig.
4H), whereas only a few neoplastic cells were positive for NF1
(Sigma-Aldrich; Merck KGaA) (Fig.
4I). Immunostaining protocol is as follows; sections were
hydrated by passage through xylene and graded ethanols. After
antigen retrieval for 10 min at 99 degree in citric buffer, pH 6.0,
the slides were blocked with 3% BSA for 1 h, then incubated with a
primary antibody for 16 h at 4 degree. After washing with PBS,
slides were mounted using ImmPRESS HRP polymer detection kit
(Vector Laboratories) and peroxidase Stain DAB kit (Brown Stain)
(Nacalai Tesque), followed by counterstaining with hematoxylin.
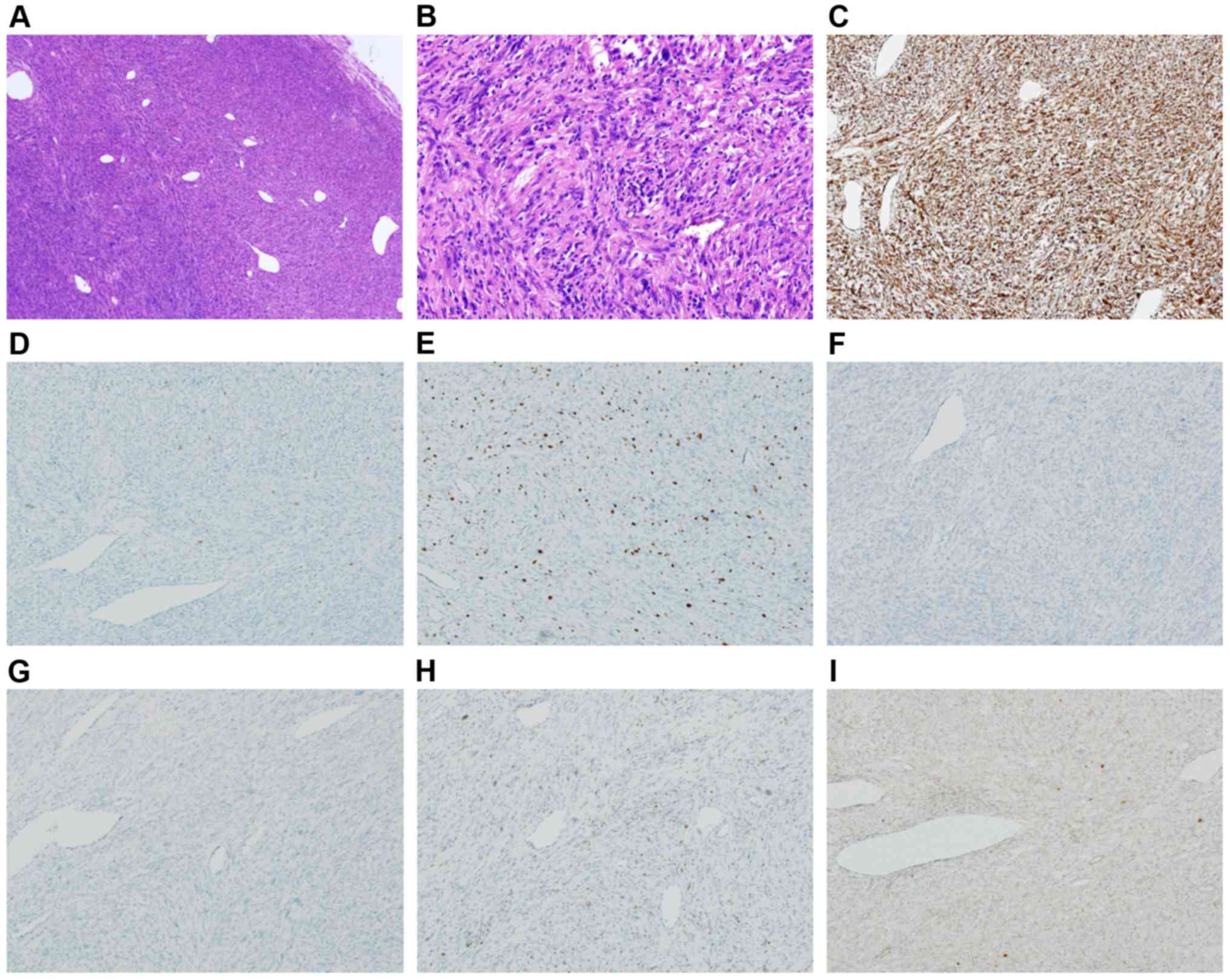 | Figure 4.Microphotographs of the surgically
removed tumor. (A) A well-circumscribed mass presenting dense
spindle-cell proliferation with hemangiopericytomatous vessels was
observed (H&E staining; magnification, x40). (B) Neoplastic
cells exhibiting moderate-to-severe nuclear atypia and focal
pleomorphism against a chronic inflammatory background (H&E
staining; magnification, x200). (C) Diffuse positivity for vimentin
(IHC staining; magnification, x100) was observed. (D) Scattered
positive cells (<1%) for S100 (IHC staining; magnification,
x100) were identified. (E) A Ki67 labeling index of 20% was
determined (IHC staining; magnification, x100). Samples were
negative for (F) SOX10 (IHC staining; magnification, x100) and (G)
CD68 (IHC staining; magnification, x100). Incomplete loss of
expression of (H) H3K27me3 (IHC staining; magnification, x100) and
(I) NF1 (IHC staining; magnification, x100). H&E, hematoxylin
and eosin; IHC, immunohistocemical. |
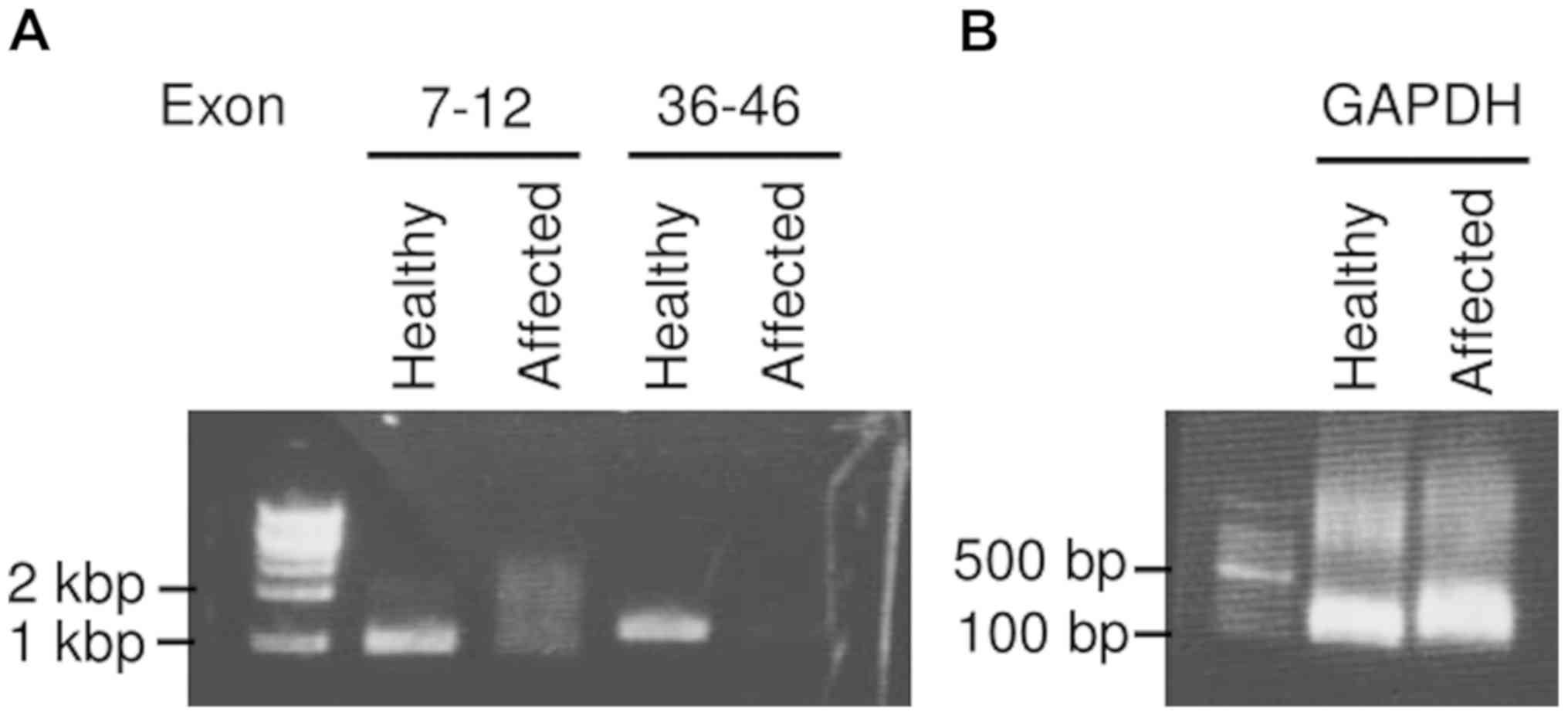
With regards to genomic mutation, reverse
transcriptase-polymerase chain reaction (RT-PCR) using the
formalin-fixed and paraffin-embedded tissues of the primary tumor
failed to detect SYT-SSX1 and SYT-SSX2 chimeric transcripts. NF1
microdeletion in the affected skin (Café-au-lait spot) was
identified via RT-PCR using PrimeSTAR HS DNA polymerase (Takara Bio
Inc.) although no mutation was observed in the healthy side. RNA
was obtained using the RNeasy mini kit (Qiagen) after tissue
homogenization and the extracted RNA was then retrotranscripted
using SuperScript IV VILO Master Mix (Thermo Fisher Scientific,
Inc.). As a DNA size marker, 1 kb or 100 bp DNA Ladder (Takara Bio
Inc.) was used. The primers used for amplifying a part of the NF1
gene were as follows: Exon 7-12: Forward
5'-AGATAACTCTGTCATTTTCCTAC-3' and reverse
5'-CTATCCATAGAGGAGTTCGCT-3' as well as exon 36-46: Forward
5'-CCAGTGGACAGAACTA-GCTC-3' and reverse
5'-GGCCTCTGCTAAGTATTCATA-3'. As an internal control, GAPDH was
measured using the following primers: Forward
5'-AGGGCTGCTTTTAACTCTGGT-3' and reverse 5'-CCCCACTTGATTTTGGAGGGA-3'
(Fig. 5).
Discussion
The etiology of NF1 is the mutation in the NF1 gene
located in chromosome 17q11.2(11).
In 2000, Tinschert et al showed that mosaic localized NF1
was caused by the somatic mutation of the NF1 gene (12).
In 1977, Miller and Sparkes reported the case of a
15-year-old girl with multiple pigmented macules, café-au-lait
spots, and a neurofibroma in a specific region of the body, which
led to the establishment of the term segmental neurofibromatosis
(4). In 1982, Riccardi proposed the
classification of NF into eight subtypes. NF5 is segmental NF,
which is defined by the limitation of café-au-lait spots, freckles,
and/or cutaneous neurofibromas to a certain region of the body
(5). In 1987, Roth et al
further classified the segmental NF into four categories: True
segmental (NF5), localized with deep involvement, hereditary
segmental, and bilateral segmental (13). In 1997, Gutmann et al proposed
the term mosaic NF1(14). In 2001,
Ruggieri and Huson further classified mosaic NF1 into two
categories: Mosaic generalized NF1 and mosaic localized
NF1(6). Based on pathogenesis, these
terms represent varying skin lesions, affected regions, and
symptoms accurately.
The prevalence rate of mosaic localized NF1 is
approximately 0.0018%, whereas that of NF1 is approximately
0.02-0.03% (9,10). Meanwhile, the incidence rate of MPNST
is 4.6% in individuals with NF1 and 0.001% in the general
population, and approximately 52% of patients with NF1 presented
with MPNST (2). Although numerous
patients with NF1 present with MPNST, only three cases involved
mosaic localized NF1 (7,8). There are two hypotheses for this
phenomenon. First, cells with gene mutation in mosaic localized NF1
are limited to the affected region and the number of cells that can
be mutate to MPNST is higher in NF1 than in mosaic localized
NF1(7). Second, mosaic localized NF1
is underdiagnosed (9). In addition,
we hypothesized that the proportion of patients with mosaic
localized NF1 who present with neurofibroma is low, although MPNST
arises in the neurofibroma of patients with NF1 in which malignant
transformation occurs. Ruggieri et al classified 124
patients with mosaic localized NF1 into four groups: Pigmentary
changes only, neurofibromas only, pigmentary changes and
neurofibromas, and isolated plexiform neurofibromas only. A total
of 86 patients were included in the pigmentary change only group,
20 in the neurofibroma only group, 10 in the pigmentary change and
neurofibroma group, and 8 in the isolated plexiform neurofibroma
only group. Pigmentary changes were often observed (6).
The resection of the tumor is the definitive
treatment for MPNST and the efficacy of adjuvant or neoadjuvant
therapy has not been fully investigated. The local recurrence rates
of resection with adequate surgical margin and with inadequate
surgical margin were 6 and 30%, respectively (2,3).
An earlier clinical report showed that the 10-year
overall survival rate of individuals with NF1-related MPNST and
sporadic MPNST are 45 and 60%, respectively, and statistically
significant differences were observed (15). The number of individuals with mosaic
localized NF1 is extremely low; thus, studies about the survival
rate of patients with MPNST arising from mosaic localized NF1 were
not conducted. In a case report, all three cases survived at least
during the follow-up period although two cases out of three caused
local recurrence (8). In the present
case, we identified NF1 microdeletion; thus, a cautious follow-up
is required not to overlook local recurrence and new lesions in the
affected leg.
The features of the histopathological findings of
MPNST are spindle cell tumors growing in storiform and positive in
immunohistochemical staining, which include S-100, CD56, and
protein gene product 9.5 (PGP 9.5). In particular, S-100 was
considered a good marker of MPNST, but it is only about 50-90%
positive (16) and there is no
immunohistochemical marker with high sensitivity and specificity
for MPNST. Previous studies have also reported on patients
diagnosed with spindle cell sarcoma associated with mosaic
localized NF1 and S-100 positive as MPNST (7,8).
Synovial sarcoma was excluded because neither SYT-SSX1 nor SYT-SSX2
fusion gene was identified via RT-PCR. Other types of sarcomas,
such as dedifferentiated liposarcoma, leiomyosarcoma,
rhabdomyosarcoma, or solitary fibrous tumor, are significantly less
common in younger patients, and their morphological and
immunohistochemical findings were inconsistent. Finally, MPNST was
the most probable diagnosis based on the immunohistochemical
analysis.
The definitive diagnosis of MPNST was challenging in
the present case. A previous report has revealed the diagnosis of
MPNST and showed that all MPNSTs are characterized by NF1 deletion,
although NF1 expression varies in plexiform neurofibroma,
indicating that the deletion of NF1 is a useful indicator of
appropriate diagnosis (17). In
addition, NF1 microdeletion was observed in both generalized and
localized NF1(18). Thus, our
patient can be diagnosed with MPNST arising from mosaic localized
NF1 due to the presence of regional café-au-lait spots and genomic
NF1 deletion.
Neurofibromatosis type 1 results from NF1
microdeletion which encompasses the entire NF1 gene. In Fig. 5, not only exon 36-46, but exon 7-12
was also deleted in the affected skin. This huge deletion of NF1
means NF1 microdeletion, which is observed only in
neurofibromatosis type 1. As far as we checked on the database of
COSMIC, NF1 microdeletion is not observed in any kinds of cancer
other than neurofibromatosis type 1. NF1 microdeletion is an
absolute causative gene (100% penetrance), not a second hit.
Earlier reports identified three patients aged
>40 years who presented with MPNST associated with mosaic
localized NF1. We reported the first case of MPNST in an adolescent
patient with mosaic localized NF1. Moreover, malignant
transformation in neurofibroma was assumed to occur during puberty
due to the fact that the exacerbation of neurofibroma during
puberty and pregnancy is well recognized (19).
In this report, MPNST was observed in an adolescent
patient with mosaic localized NF1. In the case of a challenging
histologic diagnosis, the presence of skin lesions is helpful in
the diagnosis of MPNST. In the present study, NF1 microdeletion was
consistently observed in the café-au-lait spots. MPNST associated
with mosaic localized NF1 is a rare type of malignancy. However, it
is of high-grade malignancy; therefore, orthopedists should bear in
mind this clinical feature.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HH, SN, and HT conceived the present study,
pathologically diagnosed the patient and wrote the manuscript. SN,
EK, and HT performed cytologic diagnoses, immunocytochemical
analysis and gene expression analysis. HH, TW, YI, TT, HO, NN and
HT collected clinical data. SN and HT revised the manuscript. All
authors read and approved the final manuscript prior to
submission.
Ethics approval and consent to
participate
Skin samples were collected after written informed
consent was obtained according to the protocol approved by Osaka
International Cancer Institute (Osaka, Japan). Written informed
consent was obtained from the patient and patient's parents for
study participation.
Patient consent for publication
Written informed consent was obtained from the
patient and patient's parents for the publication of patient data
and associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fletcher CDM, Unni KK and Mertens F:
Pathology and genetics of tumours of soft tissue and bone. WHO
classification of tumours. IARC Press Lyon, 2002.
|
|
2
|
Ducatman BS, Scheithauer BW, Piepgras DG,
Reiman HM and Ilstrup DM: Malignant peripheral nerve sheath tumors.
A clinicopathologic study of 120 cases. Cancer. 57:2006–2021.
1986.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Porter DE, Prasad V, Foster L, Dall GF,
Birch R and Grimer RJ: Survival in malignant peripheral nerve
sheath tumours: A comparison between sporadic and neurofibromatosis
type 1-associated tumours. Sarcoma. 2009(756395)2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Miller RM and Sparkes RS: Segmental
neurofibromatosis. Arch Dermatol. 113:837–838. 1977.
|
|
5
|
Riccardi VM: Neurofibromatosis: Clinical
heterogeneity. Curr Probl Cancer. 7:1–34. 1982.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ruggieri M and Huson SM: The clinical and
diagnostic implications of mosaicism in the neurofibromatoses.
Neurology. 56:1433–1443. 2001.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Schwarz J and Belzberg AJ: Malignant
peripheral nerve sheath tumors in the setting of segmental
neurofibromatosis. Case report. J Neurosurg. 92:342–346.
2000.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li K, Chong HW and Sang EM: A superficial
form of malignant peripheral nerve sheath tumour associated with
segmental neurofibromatosis. Acta Derm Venereol. 85:540–541.
2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ingordo V, D'Andria G, Mendicini S,
Grecucci M and Baglivo A: Segmental neurofibromatosis: Is it
uncommon or underdiagnosed? Arch Dermatol. 131:959–960.
1995.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Lammert M, Friedman JM, Kluwe L and
Mautner VF: Prevalence of neurofibromatosis 1 in German children at
elementary school enrollment. Arch Dermatol. 141:71–74.
2005.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ledbetter DH, Rich DC, O'Connell P,
Leppert M and Carey JC: Precise localization of NF1 to 17q11.2 by
balanced translocation. Am J Hum Genet. 44:20–24. 1989.PubMed/NCBI
|
|
12
|
Tinschert S, Naumann I, Stegmann E, Buske
A, Kaufmann D, Thiel G and Jenne DE: Segmental neurofibromatosis is
caused by somatic mutation of the neurofibromatosis type 1 (NF1)
gene. Eur J Hum Genet. 8:455–459. 2000.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Roth RR, Martines R and James WD:
Segmental neurofibromatosis. Arch Dermatol. 123:917–920.
1987.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Gutmann DH, Aylsworth A, Carey JC, Korf B,
Marks J, Pyeritz RE, Rubenstein A and Viskochil D: The diagnostic
evaluation and multidisciplinary management of neurofibromatosis 1
and neurofibromatosis 2. JAMA. 278:51–57. 1997.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hwang IK, Hahn SM, Kim HS, Kim SK, Kim HS,
Shin KH, Suh CO, Lyu CJ and Han JW: Outcomes of treatment for
malignant peripheral nerve sheath tumors: Different clinical
features associated with neurofibromatosis type 1. Cancer Res
Treat. 49:717–726. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Stasik CJ and Tawfik O: Malignant
peripheral nerve sheath tumor with rhabdomyosarcomatous
differentiation (malignant triton tumor). Arch Pathol Lab Med.
130:1878–1881. 2006.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Perry A, Roth KA, Banerjee R, Fuller CE
and Gutmann DH: NF1 deletions in S-100 protein-positive and
negative cells of sporadic and neurofibromatosis 1 (NF1)-associated
plexiform neurofibromas and malignant peripheral nerve sheath
tumors. Am J Pathol. 159:57–61. 2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Messiaen L, Vogt J, Bengesser K, Fu C,
Mikhail F, Serra E, Garcia-Linares C, Cooper DN, Lazaro C and
Kehrer-Sawatzki H: Mosaic type-1 NF1 microdeletions as a cause of
both generalized and segmental neurofibromatosis type-1 (NF1). Hum
Mutat. 32:213–219. 2011.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Riccardi VM: Von Recklinghausen
neurofibromatosis. N Engl J Med. 305:1617–1627. 1981.PubMed/NCBI View Article : Google Scholar
|