Introduction
Colorectal cancer (CRC) is the 3rd and 4th most
commonly diagnosed cancer globally in women and men, respectively
(1-3).
The transformation of the normal colonic epithelium is the result
of the progressive accumulation of genetic and epigenetic
alterations that promote tumor growth. Colon cancer cells result
from a multi-step progression in molecular and morphological
changes of normal cells. In addition, genetic instability and germ
line genetic defects drive the initiation of sporadic colon cancers
(4-6).
The RAS oncogene family, comprising three known
human isoforms, NRAS, HRAS and KRAS, regulates a number of cellular
functions, including cell proliferation, apoptosis, migration and
differentiation (7). KRAS mutations
comprise 86% of all RAS mutations in human cancers (8). The frequency of KRAS mutations is the
highest (21.6%) in all cancers, followed by NRAS (8.0%) and HRAS
(3.3%) (9). The hotspot of KRAS
mutations is located in exon 2, and the most frequent changes are
identified in codons 12 (~82% of all reported KRAS mutations) and
13 (~17%). Other mutations of KRAS have been detected in codons 61
and 146, but these have little or no effect on the progression of
CRC (10). The most frequent change
is the transition of GGT to GAT in codon 12, specifically DNA
nucleotide mutations involving G→A or G→T (7). However, 60% of NRAS mutations occur
mainly in codon 61 vs. 35% in codon 12, while HRAS mutations in CRC
show an intermediate pattern with an approximate 50:40 split
between mutations in codons 12 and 61, respectively (11). KRAS and HRAS mutations comprise
single-nucleotide point mutations, with the most common
substitutions in KRAS resulting in G12D, G12A, G12R, G12C, G12S,
G12V and G13D substitutions and the most frequent codon 12
substitutions being G12D and G12V (10). However, in HRAS, the most common
mutation results in a G12V substitution (11). The presence of a glycine residue in
codon 12, therefore, plays an important role in the normal function
of the RAS proteins. This single-nucleotide substitution that
results in the replacement of glycine with another amino acid leads
to the formation of a constitutively active GTPase (12).
RAS proteins are activated when nearby transmembrane
receptors, such as growth factor receptors, G-protein coupled
receptors and toll-like receptors, are bound by the corresponding
ligand. The subsequent intracellular signaling cascade involves
guanine exchange factors (GEF) that facilitate the activation of
RAS by replacing the inactive GDP with GTP. RAS activation triggers
the downstream activation of a wide variety of effectors, including
serine/threonine kinases, GTPase-activating proteins (GAPs),
phosphoinositide 3-kinase and GEFs. RAS is deactivated when the GTP
is hydrolyzed to GDP. When GTP binds to the KRAS protein, KRAS
undergoes a conformational change involving two regions of the
protein known as Switch 1 (amino acids 30-38) and Switch 2 (amino
acids 59-67), which form an effector loop controlling the
specificity of the binding of this GTPase to its effector
molecules. The changes in KRAS conformation affect its interactions
with multiple downstream transducers, such as GAPs, which amplify
the GTPase activity of RAS 100,000-fold (13). Once KRAS is mutated, it becomes
constitutively active and the regulation of downstream functions is
lost, resulting in unregulated cell growth (7). Screening of KRAS mutations is widely
used in clinical practice to decide on the treatment for CRC
(14). Recently, KRAS mutation
screening has become common practice before prescribing
anti-epidermal growth factor receptor monoclonal antibodies in the
treatment of advanced CRC (15).
CRC is the second most common type of cancer among
patients admitted to the National Cancer Institute (NCI)-Misurata,
where ~180 new cases (52% male and 48% female patients) were
admitted in 2019. The incidence and mortality rates for CRC
increase with age, with >90% of the patients aged >50 years
(2). The aim of the present study
was to investigate the presence of KRAS and HRAS mutations among
patients with CRC in NCI-Misurata, in order to determine whether
the frequency of those mutations is associated with the type and
stage of CRC.
Materials and methods
Tissue samples
The present study included 34 frozen tumor tissue
samples obtained from patients with CRC, who underwent curative
surgery at NCI-Misurata. The patients were diagnosed at the
Department of Histopathology between December 2016 and August 2017.
The study protocol was approved by the Ethics Committee of
NCI-Misurata. All tumor tissues were placed on ice immediately upon
removal from the patient, sectioned and stored at -80˚C until DNA
was extracted.
Clinical records
The clinical and demographic data collected from
patient records included: Age, sex, family history of CRC, tumor
site, degree of tumor differentiation, tumor stage and histological
type, such as common adenocarcinoma or mucinous carcinoma. Patients
with family history of CRC were selected for the present study. All
the patients have signed a consent form prior to the use of the
studied tissues, according to the regulations of the
NCI-Misurata.
DNA extraction and PCR reaction
Genomic DNA extraction from frozen tumor tissues was
carried out using QIAamp DSP DNA Fresh Tissue kit (Qiagen GmbH).
After the washing steps, the DNA was eluted in Tris/EDTA buffer and
stored at -20˚C. The DNA was quantitated using a NanoDrop
spectrophotometer (Thermo Fisher Scientific, Inc.). After
extraction of the genomic DNA from the samples, the KRAS and HRAS
exons 1and 2 were amplified using a PCR SimpliAmp™ Thermal Cycler
(Thermo Fisher Scientific, Inc.) using the forward
3'-AAGGCCTGCTGAAAATGACTG-5' and reverse 3'-CAAAGAATGGTCCTGCACCAG-5'
primers for KRAS-exon 2 and the forward 3'-GGGCCCTCCTTGGCAGGTGG-5'
and reverse 3'-CACCTGGACGGCGGCGCCAG-5' primers for HRAS-exon 2.
Each PCR reaction was performed in a volume of 20 µl containing PCR
buffer, 2.5 mM MgCl2, 5 units of Taq polymerase, 1 mM of
each dNTP, forward and reverse primers (10 pmol) and 100 ng of
genomic DNA. The PCR reaction ran with the following program: 95˚C
initial denaturation for activating the Fast Star Taq DNA
Polymerase for 5 min; step 2: Denaturation at 95˚C for 30 sec; step
3: Annealing of the primers to the template at 54˚C for HRAS in
exons 1 and 2, 45˚C for KRAS in exon 1, and 43˚C for exon 2 for 30
sec; and step 4: 3 min at 72˚C for primer extension. Steps 2-4 were
repeated 25 times, followed by a final extension step at 72˚C for 5
min; subsequently, the PCR reaction was cooled down to 8˚C. The PCR
products were subjected to direct DNA sequencing.
The amplification product sizes were confirmed by
electrophoresis on 1% agarose gel in Tris-borate-EDTA buffer
(Sigma-Aldrich; Merck KGaA), and visualized on a UVP BioSpectrum™
500 Imaging System (VWR Corporation). The PCR reaction products
were purified using the MinElute PCR Purification Kit (Qiagen GmbH)
and subjected to sequence analysis.
Direct DNA sequencing
Direct sequencing of PCR products was performed
after purification with the PCR Product Purification Kit (Roche
Diagnostics GmbH). The forward primers used in PCR amplification of
KRAS and HRAS genes were used for sequencing in on the EVO150
Genetic Analyzer (Tecan Group, Ltd./Applied Biosystems; Thermo
Fisher Scientific, Inc.). To ensure the accuracy of sequencing
results, all the molecular tests and the direct sequencing analysis
were performed twice on each sample. All DNA sequencing reactions
were performed by Inqaba Biotec.
Statistical analysis
Data analysis was performed using SPSS version 17.0
(SPSS Inc.). The Fisher's exact test was used for comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
KRAS and HRAS mutations in
histopathological biopsies
KRAS mutations were identified in 13/34 (38.2%) of
the tumors, while all the patients in this study group had the
wild-type HRAS gene (Fig. 1). The
site of the tumor was found to be associated with KRAS mutations
(P=0.027). Tumors located in the left colon had a significantly
higher frequency of mutant KRAS status (8/11; 72.7%) compared with
those located in the right colon. Table
I shows the frequency and association with clinicopathological
characteristics of patients who harbored mutant and wild-type KRAS
tumors; 76.9% (10/13) of the patients with tumors harboring mutant
KRAS presented with stage B, C or D colon cancer, indicating that
patients with mutant KRAS tumors are more likely to have
advanced-stage disease. Tumor differentiation status, sex, patient
age and stage at diagnosis were not identified as significant
predictors of KRAS status (P=0.458).
 | Table IDistribution of tumour characteristics
according to KRAS status. |
Table I
Distribution of tumour characteristics
according to KRAS status.
| | KRAS mutation
status | |
|---|
| Characteristics | Overall | Mutant | Wild-type | P-value |
|---|
| Number of
patients | 34 | 13 | 21 | |
| Sex | | | | 0.107 |
|
Male | 19 | 5 | 14 | |
|
Female | 15 | 8 | 7 | |
| Age (years) | | | | 0.524 |
|
≤50 | 10 | 3 | 7 | |
|
>50 | 24 | 10 | 14 | |
| Localization | | | | 0.027 |
|
Right
colon | 11 | 3 | 8 | |
|
Left
colon | 11 | 8 | 3 | |
|
Rectosigmoid | 4 | 0 | 4 | |
|
Rectum | 8 | 2 | 6 | |
| Differentiation | | | | 0.458 |
|
High | 16 | 8 | 8 | |
|
Moderate | 12 | 4 | 8 | |
|
Low | 2 | 0 | 2 | |
|
Unknown | 4 | 1 | 3 | |
| Duke's stage | | | | 0.561 |
|
A | 3 | 2 | 1 | |
|
B | 7 | 4 | 3 | |
|
C | 18 | 5 | 13 | |
|
D | 3 | 1 | 2 | |
|
Unknown | 3 | 1 | 2 | |
Type and frequency of KRAS gene
mutations
The type and frequency of KRAS gene mutations are
detailed in Table II. Of the 13
tumors, 12 (92.3%) had mutations in codon 12, while 1 had a
mutation in codon 13 (7.7%). Of the 13 patients with CRC harboring
KRAS mutations 6 (46.1%) had a Gly12Asp mutation 4 (30.8%) had a
Gly12Val mutation, 2 (15.4%) had a Gly12Cys mutation, and 1 (7.7%)
had a Gly13Asp mutation. Mutations from G to A occurred in 7/13
(53.8%) of the patients, and from G to T in 6/13 (46.2%) of the
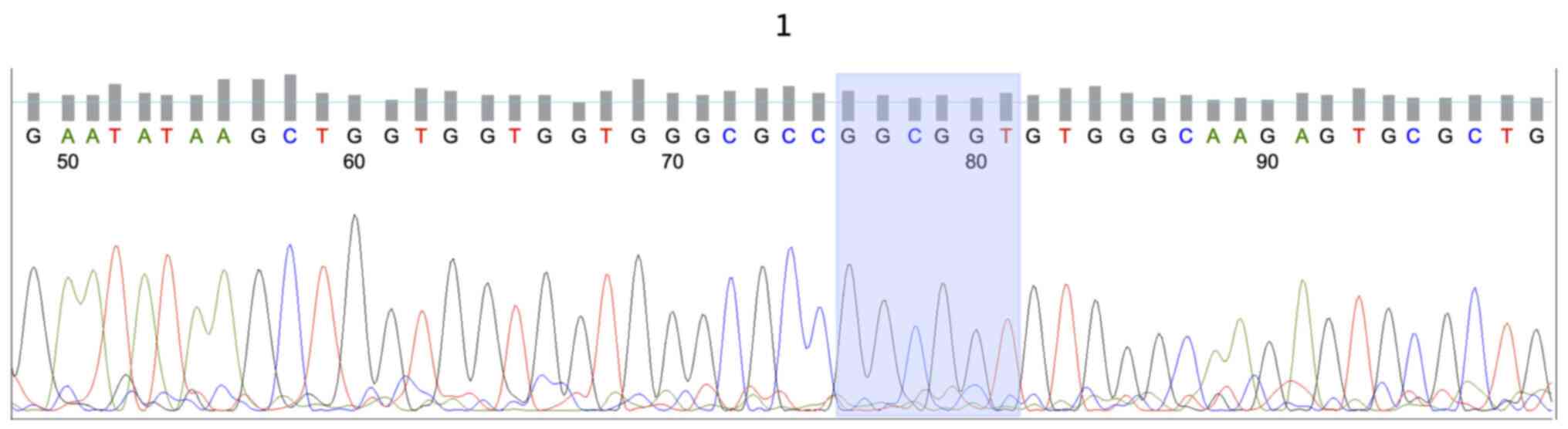
patients (Fig. 2). Mutations
occurred at the first base of codon 12 or 13 in 2/13 (15.4%) and at
the second base in 11/13 (84.6%) of the patients (Table II).
| Table IIDistribution of KRAS mutation
types. |
Table II
Distribution of KRAS mutation
types.
| Codon | Amino acid
substitution | Incidence, % | Number of
samples | Base change |
|---|
| 12 | Gly12Asp | 46.1 | 6 | GGT>GAT |
| 12 | Gly12Val | 30.8 | 4 | GGT>GTT |
| 12 | Gly12Cys | 15.4 | 2 | GGT>TGT |
| 13 | Gly13Asp | 7.7 | 1 | GGC>GAC |
Discussion
The present study investigated the association
between clinicopathological factors and KRAS mutation status in
Libyan patients with CRC and, except for the tumor site, no other
significant associations were identified. This observation is
consistent with the findings of a Saudi Arabian study (16) and the RASCAL multicenter study
(17), which demonstrated a trend
for higher mutation frequency in tumors of the left colon (51 and
58.8%, respectively) compared to other sites (16,17).
This finding has been attributed to the increased exposure of the
left-sided bowel lumen to ingested carcinogens and mutagens. By
contrast, KRAS mutations have been reported to be slightly more
frequent in the right colon compared with other sites (47 vs. 41%,
respectively) in studies from Saudi Arabia (18,19),
while another study from Turkey reported that the KRAS mutation
frequency was significantly higher in tumors located in the
ascending colon (20).
There are several factors that may explain the
variations in the observed frequencies of KRAS mutations according
to tumor location in different studies. These include the use of
different methodologies for the detection of mutations, with
different ranges of sensitivity and specificity. Environmental
factors have a major impact on different populations with diverse
lifestyles, dietary habits, and variable exposures to carcinogens.
Furthermore, the genetic polymorphisms in the
carcinogen-metabolizing genes in different population groups must
also be taken into consideration (21-24).
KRAS mutations were identified in 38.2% of all CRC
patients in our study group. This finding was in agreement with
previous reports that identified KRAS mutations in 30-40% of
patients with CRC (17,21). Consistent with previous reports,
92.3% of the studied tumors had a single base mutation at codon 12
of the KRAS gene, and 7.7% had a single base mutation at codon 13
(17,21,22).
The most common single base mutation (46.1%) was a GGT to a GAT
transition in codon 12 that resulted in a change from the amino
acid glycine to aspartic acid (Gly12Asp). The frequency of KRAS
mutations in our study group did not differ when compared with
those of most other studies in Europe and the United States
(23,24). Previous studies that enrolled a
relatively large number of Arabian patients with different stages
of the disease, reported KRAS mutation rates of 28-56% in Saudi
Arabia (19,25-27),
23-32% in Tunisia (22,28,29),
33-44% in Jordan (30,31) and 42% in Egypt (32).
A number of factors may be associated with
variations in the observed frequencies of KRAS mutations in
different studies. Among these, environmental factors may be the
most important parameter that may explain the difference in KRAS
mutations between Libya and neighboring countries, such as Tunisia
and Egypt, and western countries.
In conclusion, KRAS genetic alterations in codons 12
and 13 were found to be higher in this group of Libyan patients
with CRC. However, further studies are required to fully elucidate
the molecular background of CRC, by investigating other genes, such
as APC, BRAF and NRAS, as well as other exons in the KRAS, HRAS and
NRAS genes. Future studies of larger patient groups may provide
more accurate information regarding the association between KRAS,
HRAS and NRAS mutations and the clinicopathological characteristics
of patients with CRC.
Acknowledgements
We would like to thank Dr Fatima Maetig and Dr
Yachine Topov from NCI-Misurata for their pathological
re-examinations of the CRC biopsies. We thank Dr Mohamed Elfagieh
and the Scientific Committee of NCI-Misurata for their support for
this project. We thank all the staff in the Parker Laboratory for
their assistance during the project. We are grateful to Dr Alhusen
Saad and Mr. Ahmed Hamouda from the Faculty of Engineering,
Misurata University, for image processing of the sequencing.
Funding
MIP was supported by the SMART fellowship from the ICGEB, and
research grants the South African Medical Research Council and the
University of Cape Town.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AA conducted the experiments and the data analysis.
MD assisted with the analysis of the obtained results. MA provided
the clinical data of the patients with CRC. MIP was the
co-supervisor of the study and critically revised the manuscript
for important intellectual content. OA supervised the project,
experimental design and data analysis, and edited the manuscript.
MIP and OA have assessed and confirmed the authenticity of raw
data. All the authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were in accordance with the ethical standards of the
National Cancer Institute-Misurata, the institutional research
committee, and with the 1964 Helsinki Declaration and its later
amendments or comparable ethical standards. All patients have
signed the consent prior to the use of the studied tissues
according to the regulations of the NCI-Misurata.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Al-Sohaily S, Biankin A, Leong R,
Kohonen-Corish M and Warusavitarne J: Molecular pathways in
colorectal cancer. J Gastroenterol Hepatol. 27:1423–1431.
2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ivanovich JL, Read TE, Ciske DJ, Kodner IJ
and Whelan AJ: A practical approach to familial and hereditary
colorectal cancer. Am J Med. 107:68–77. 1999.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumourigenesis. Cell. 61:759–767.
1990.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649.
1998.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Vogelstein B and Kinzler KW: Lessons from
hereditary colorectal cancer. Cell. 87:159–170. 1996.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Arrington A, Heinrich E, Lee W, Duldulao
M, Patel S, Sanchez J, Garcia-Aguilar J and Kim J: Prognostic and
predictive roles of KRAS mutation in colorectal cancer. Int J Mol
Sci. 13:12153–12168. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Baines AT, Xu D and Der CJ: Inhibition of
ras for cancer treatment: The search continues. Future Med Chem.
3:1787–1808. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bamford S, Dawson E, Forbes S, Clements J,
Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR
and Wooster R: The COSMIC (Catalogue of Somatic Mutations in
Cancer) database and website. Br J Cancer. 91:355–358.
2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Tan C and Du X: KRAS mutation testing in
metastatic colorectal cancer. World J Gastroenterol. 18:5171–5180.
2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Prior IA, Lewis PD and Mattos C: A
comprehensive survey of ras mutations in cancer. Cancer Res.
72:2457–2467. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Jančík S, Drábek J, Radzioch D and Hajdúch
M: Clinical relevance of KRAS in human cancers. J Biomed
Biotechmol. 2010(150960)2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Gideon P, John J, Frech M, Lautwein A,
Clark R, Scheffler JE and Wittinghofer A: Mutational and kinetic
analyses of the GTPase-activating protein (GAP)-p21 interaction:
The C-terminal domain of GAP is not sufficient for full activity.
Mol Cell Biol. 12:2050–2056. 1992.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kinzler K and Vogelstein B: The genetic
basis of cancer. In: Cancer Gene Therapy. Curiel DT and Douglas JT
(eds). Humana Press, Totowa, pp9-18, 2002.
|
|
15
|
Lee S, Brophy VH, Cao J, Velez M, Hoeppner
C, Soviero S and Lawrence HJ: Analytical performance of a PCR assay
for the detection of KRAS mutations (codons 12/13 and 61) in
formalin-fixed paraffin-embedded tissue samples of colorectal
carcinoma. Virchows Arch. 460:141–149. 2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bader T and Ismail A: Higher prevalence of
KRAS mutations in colorectal cancer in Saudi Arabia: Propensity for
lung metastasis. Alexandria J Med. 50:203–209. 2014.
|
|
17
|
Andreyev HJ, Norman AR, Clarke PA,
Cunningham D and Oates J: Kirsten ras mutations in patients with
colorectal cancer: The multicenter RASCAL study. J Nat Cancer Inst.
90:675–684. 1998.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zekri J, Rizvi A, Al-Maghrabi J and Bin
Sadiq B: K-Ras in colorectal cancer tumors from Saudi patients:
Frequency, clinicopathological association and clinical outcome.
Open Colorectal Cancer J. 5:22–27. 2012.
|
|
19
|
Abubaker J, Bavi P, Al-Haqawi W, Sultana
M, Al-Harbi S, Al-Sanea N, Abduljabbar A, Ashari LH, Alhomoud S,
Al-Daye F, et al: Prognostic significance of alterations in KRAS
isoforms KRAS-4A/4B and KRAS mutations in colorectal carcinoma. J
Pathol. 219:435–445. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Gorukmez O, Yakut T, Gorukmez O, Sag SO,
Karkucak M and Kanat O: Distribution of KRAS and BRAF mutations in
metastatic colorectal cancers in Turkish patients. Asian Pac J
Cancer Prev. 17:1175–1179. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Smith G, Carey FA, Beattie J, Wilkie MJ,
Lightfoot TJ, Coxhead J, Garner RC, Steele RJ and Wolf C: Mutations
in APC, Kirsten-ras, and p53-alternative genetic pathways to
colorectal cancer. Proc Natl Acad Sci. 99:9433–9438.
2002.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Aissi S, Buisine M, Zerimech F, Kourda N,
Moussa A, Manai M and Porchet N: KRAS mutations in colorectal
cancer from Tunisia: Relationships with clinicopathological
variables and data on TP53 mutations and microsatellite
instability. Mol Biol Rep. 40:6107–6112. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Edkins S, O'Meara S, Parker A, Stevens C,
Reis M, Jones S, Greenman C, Davies H, Dalgliesh G, Forbes S,
Hunter C, et al: Recurrent KRAS codon 146 mutations in human
colorectal cancer. Cancer Biol Ther. 5:928–932. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Licar A, Cerkovnik P, Ocvirk J and
Novakovic S: KRAS mutations in Slovene patients with colorectal
cancer: Frequency, distribution and correlation with the response
to treatment. Int J Oncol. 36:1137–1144. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zekri J, Al-Shehri A, Mahrous M,
Al-Rehaily S, Darwish T, Bassi S, El Taani H, Al Zahrani A,
Elsamany S, Al-Maghrabi J and Sadiq BB: Mutations in codons 12 and
13 of K-ras exon 2 in colorectal tumors of Saudi Arabian patients:
Frequency, clinicopathological associations, and clinical outcomes.
Genet Mol Res. 16(4238)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Beg S, Siraj A, Prabhakaran S, Bu R,
Al-Rasheed M, Sultana M, Qadri Z, Al-Assiri M, Sairafi R, Al-Sanea
N, et al: Molecular markers and pathway analysis of colorectal
carcinoma in the Middle East. Cancer. 121:3799–3808.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dallol A, Buhmeida A, Al-Ahwal MS,
Al-Maghrabi J, Bajouh O, Al-Khayyat S, Alam R, Abusanad A, Turki R,
Elaimi A, et al: Clinical significance of frequent somatic
mutations detected by high-throughput targeted sequencing in
archived colorectal cancer samples. J Transl Med.
14(118)2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Sammoud S, Khiari M, Semeh A, Amine L,
Ines C, Amira A, Lilia K, Taher K, Sabeh M and Saadia B:
Relationship between expression of ras p21 oncoprotein and mutation
status of the K-ras gene in sporadic colorectal cancer patients in
Tunisia. Applied Immunohistochem Mol Morphol. 20:146–152.
2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Aissi S, Buisine M, Zerimech F, Kourda N,
Moussa A, Manai M and Porchet N: Somatic molecular changes and
histopathological features of colorectal cancer in Tunisia. World J
Gastroenterol. 19:5286–5294. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Elbjeirami W and Sughayer M: KRAS
mutations and subtyping in colorectal cancer in Jordanian patients.
Oncol Lett. 4:705–710. 2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gumus M, Dane F, Karabulut B, Uygun K,
Orhan B, Aydin K and Ozen R: Interim results of observational study
to determine K-ras mutation rates in patients with metastatic
colorectal cancer in Turkey. Eur J Cancer. 49 (Suppl):S571.
2013.
|
|
32
|
Elsabah M and Adel I: Immunohistochemical
assay for detection of K-ras protein expression in metastatic
colorectal cancer. J Egypt Nat Cancer Inst. 25:51–56.
2013.PubMed/NCBI View Article : Google Scholar
|