Parkinson's disease (PD) is the second most common
neurodegenerative disorder following Alzheimer's disease (AD). It
affects an estimated 7 to 10 million individuals worldwide, a
number that is projected to double by the year 2030 due to the
aging of the population. The prevalence varies, ranging from 41
individuals per 100,000 individuals in the fourth decade of life to
>1,900 per 100,000 individuals among those aged ≥80 years. The
incidence, or the rate of newly diagnosed cases, tends to increase
with age, although it may stabilize in individuals >80 years of
age (https://parkinsonsnewstoday.com/parkinsons-disease-statistics/).
PD presents a diverse range of motor and non-motor
symptoms. The main non-motor symptoms are neuropsychiatric
features, insomnia, excessive daytime sleepiness and autonomic
dysfunction. Anxiety and depression are common neuropsychiatric
symptoms in PD, occurring from the early pre-motor phase to the
advanced stages of the disease. These symptoms vary in intensity
depending on the motor state, with anxiety being particularly
prominent during ‘off’ ‘periods. Anxiety and depression often
coexist, and it is crucial to identify the specific anxious
depressive phenotype to effectively manage both conditions
(1). Cognitive decline and dementia
are typically regarded as a part of late-stage PD or as a result of
aging; as many as 83% of individuals with PD may experience a
certain degree of cognitive dysfunction (2). PD dementia (PDD) is associated with a
marked decrease in cortical cholinergic activity. This decrease
helps to explain why certain patients may have a limited clinical
response to cholinesterase inhibitors (3). Early-stage PD often includes mild
cognitive impairment (MCI), which, due to subjective cognitive
decline, can be overlooked in clinical practice. The primary
characteristic of this cognitive syndrome is a decline in executive
function (4). Early cognitive
impairment is classified as a frontostriatal condition that relies
on dopamine and can be treated by dopaminergic medications,
particularly concerning executive function (5). The majority of individuals with PD
experience disruptions in their sleep and wakefulness, and the
frequency of these disruptions tends to increase as the disease
progresses over time (6). These
abnormalities manifest via diverse mechanisms. Daytime drowsiness
and sudden episodes of falling asleep can be distinguished from
sleep problems that occur during the night. Nocturnal sleep
disorders encompass several conditions, such as insomnia, which can
be caused by an illness or medication and involve disrupted sleep
and frequent, lengthy periods of waking up. Other disorders include
rapid eye movement behavior disorder (RBD), periodic limb
movements, restless leg syndrome and akathisia (7).
Autonomic dysfunction is a frequent occurrence in PD
and can occur prior to the appearance of motor symptoms. However,
its prevalence increases as the disease advances. Autonomic issues
involve failure in the bladder, bowel, and sexual functions, as
well as cardiovascular complications, such as postural hypotension
(8,9). Urinary dysfunction in PD encompasses
symptoms, such as waking up at night to urinate (nocturia) and
experiencing a higher frequency and urgency to urinate, which are
linked to an overactive bladder muscle (detrusor hyperreflexia).
The regulation of micturition relies on the autonomic arc of the
sacral spinal cord segments. However, it is consistently
facilitated by the pontine micturition center. The storage
function, on the other hand, is facilitated by the hypothalamus,
cerebellum, frontal cortex and basal ganglia (10-12).
Bladder hyperreflexia in PD is considered to be associated with the
absence of the inhibitory function of the basal ganglia. Imaging
studies have revealed decreased dopaminergic function and an
increased activity in the globus pallidus in individuals with PD
with bladder dysfunction, as compared to patients with PD with
normal bladder function (13,14).
Gastrointestinal dysfunction is present across the entire
gastrointestinal tract in PD, manifesting as excessive salivation,
difficulty swallowing (dysphagia), delayed stomach emptying
(impaired gastric emptying), constipation, and difficulty with
bowel movements (impaired defecation) (15). The etiology of gastrointestinal
dysfunction remains poorly comprehended and may encompass both
external and intrinsic clinical and physiological alterations.
Various neurotransmitters and neuromodulators regulate bowel
function, such as acetylcholine, 5-HT, dopamine, noradrenaline,
vasoactive intestinal peptide and nitric oxide (16). The cardiovascular system is
significantly affected in PD, with various autonomic dysfunctions
playing a crucial role. The heart is controlled by autonomic fibers
that provide innervation, specifically sympathetic (noradrenergic
and adrenergic) and parasympathetic (cholinergic) fibers. These
fibers regulate both the heart rate and the force of its
contractions. Up to 80% of individuals diagnosed with PD may
exhibit cardiac autonomic dysfunction (17). This condition includes orthostatic
hypotension (OH) and labile hypertension. OH is a sign of impaired
sympathetic function and is frequently observed in PD, with a
reported occurrence rate of 30-58% (18). Research has demonstrated that
individuals with supine hypertension (SH) in PD are more likely to
have end-organ damage, as well as an elevated risk of stroke and
cardiovascular events (19). Sensory
abnormalities are commonly reported, and a significant number of
patients suffer pain throughout the progression of their condition
(20,21). Various factors have been identified
as the causes of pain, although a primary ‘central’ pain is also
well-known (22). Prior clinical
research has established that the basal ganglia plays a crucial
role in sensorimotor functioning and action selection, both of
which necessitate the integration of multisensory information
(23). Postural instability is
commonly attributed to disrupted motor programming in the basal
ganglia. The basal ganglia directly affect the automatic control of
postural muscle tone and postural reflexes by connecting them with
the brainstem (24). A summary of
the main non-motor symptoms is presented Fig. 1.
PD is a complex, progressive neurodegenerative
disorder, and the presentation of the disease in each patient is
unique (25). The severe depletion
of dopaminergic neurons of the nigrostriatal system characterizes
and likely produces the characteristic movement disorders (resting
tremor, bradykinesia, rigidity and postural instability) in PD
(26). However, the pathophysiology
of PD involves dysfunction, not only in the dopaminergic system,
but also in the cholinergic, serotonergic and noradrenergic
systems. Therefore, as PD progresses, common non-motor symptoms,
such as cognitive decline, sleep disturbances, mood disorders and
gastrointestinal, genitourinary, or cardiovascular issues become
more pronounced (27).
The origin of blood pressure (BP) abnormalities in
patients with PD is multifactorial. It involves neurogenic factors
related to PD pathophysiology, and both peripheral and central
denervation. Additionally, treatment plays a role, as almost all
dopaminergic medications (such as levodopa and dopamine agonists)
can lead to decreased BP (28).
Cognitive deficits manifest as impairments in executive functions,
including planning, working memory and visuospatial attention. Over
time, these deficits may evolve into clinically significant
dementia. Even MCI has been identified as a predictor of declining
functional outcomes (29).
Emerging evidence indicates a potential association
between cardiovascular autonomic dysfunction and cognitive
impairment in PD (30).
Specifically, OH is associated with PDD (18). OH results from a dysfunction in the
sympathetic noradrenergic system and is clinically significant in
20-50% of patients with PD (31).
The present narrative review aimed to provide an overview of the
current understanding of the connection between cardiovascular
dysautonomia (focusing on OH, SH and non-dipping effect) and
cognitive function in PD.
OH can significantly affect individuals with PD,
particularly during the later stages of the disease. OH is
characterized by a decrease in systolic BP of at least 20 mmHg or a
reduction in diastolic BP of at least 10 mmHg when standing or
tilting the head up to at least 60˚ within a period of 3 min
(32). Patients with recumbent
hypertension should exhibit a decrease in systolic BP of at least
30 mmHg (33). The symptoms of OH,
such as frequent fainting, lightheadedness, fatigue, nausea,
trembling, headache, or pain in the neck and shoulder area, known
as ‘coat-hanger pain’, may become more severe in the morning, after
physical activity in hot environments, in the setting of
dehydration, or after consuming alcohol. Symptomatic OH can cause
significant disability and an increased risk of dangerous falls
(34). In a previous study, syncope,
which is a temporary loss of consciousness, was found in 4% of
patients with PD and in 1% of the control group (35). The frequency of these symptoms
increases as the disease advances (36). Screening for neurogenic OH (nOH)
using orthostatic symptom questionnaires, orthostatic BP
measurements and specialized autonomic testing may be beneficial
for the identification of symptomatic and asymptomatic cases as
cardiac sympathetic denervation and nOH can occur even during the
early (premotor) stages of PD (37).
One of the scales most frequently used in research for grading OH
in patients with PD is the orthostatic grading scale (38).
Patients diagnosed with OH may also have SH, which
is characterized by a systolic BP ≥150 mmHg or a diastolic BP ≥90
mmHg (39). SH typically does not
cause any noticeable symptoms. However, some individuals may
experience a throbbing headache when lying flat. It has the
potential to cause ventricular hypertrophy (40), renal impairment (41) and intracranial hemorrhage (42).
As per the American Heart Association Council on
High BP Research, the European Society of Hypertension, and the
Japanese Society of Hypertension, nocturnal BP is considered normal
if the average nighttime values are <120/70 mmHg. However,
values >125/75 mmHg are considered abnormal (43-45).
In a previous study, the nocturnal BP of patients who experienced a
lack of sleep of at least 2 h lost its predictive value following a
minimum of 7 years of monitoring for mortality and overall
cardiovascular risk (46). However,
in another study, the association between nocturnal BP levels and
cardiac hypertrophy appeared less prominent when sleep was
disrupted during overnight BP monitoring (47). Assembly data indicate that the
measurement of nocturnal BP is a more accurate predictor of
cardiovascular disease outcomes than daytime or 24-h BP
measurements. As a result, nocturnal BP measurement is gaining
significance in clinical practice (48-51).
The clinical implications of nocturnal BP vs. daytime BP are more
pronounced in treated hypertensive patients, as demonstrated by
previous a meta-analysis of the International Database on
Ambulatory BP Concerning Cardiovascular Outcome. This analysis
included 7,458 participants with a mean age of 57 years. The
findings indicated that daytime BP was a significant predictor of
cardiovascular events in treated hypertensive patients after
adjusting for nighttime BP (52).
Nocturnal BP dipping refers to the normal reduction in nocturnal BP
compared to daytime BP. While it may seem random, a 10-20%
reduction in nighttime BP compared to daytime BP is generally
considered normal. The term ‘non-dippers’ is used to refer to a
specific group of patients with hypertension who experience a
nocturnal BP decline <10/5 mmHg and have a high risk of stroke
(53). Individuals classified as
non-dippers have a higher prevalence of heart hypertrophy, silent
cerebral infarction and microalbuminuria compared to those
classified as dippers (54-56).
Postprandial hypotension (PPH) is characterized by a
decrease in systolic BP ≥20 mmHg within a time frame of 2 h after
consuming a meal (57). Food
ingestion does not typically affect systemic BP in healthy
individuals. However, alterations in gastrointestinal and
pancreatic hormones trigger compensatory reactions in the heart and
regional blood flow. Prior research conducted on individuals with
autonomic failure has demonstrated that PPH can occur even when
lying down due to the defective compensatory mechanisms for
postprandial splanchnic blood pooling. Meals rich in carbohydrates
are more likely to reduce BP than meals rich in protein or fat. PPH
exacerbates OH (58). Studies have
provided conflicting results as regards the degree of PPH in PD.
Untreated PD has been shown to be associated with a slight decrease
in BP after eating while lying down (59). It has been observed that older
patients with PD experience a higher occurrence and severity of PPH
compared to OH (60).
Post-exercise hypotension (PEH) is the temporary
decrease in BP following a single exercise session. There are no
specific standards for determining the extent and duration of the
decline in BP after exercise that can be used to diagnose PEH
(61). Patients with autonomic
failure experience a decrease in BP and a worsening of OH when
exercising, even in a supine position (62). A consistent reduction in systolic and
diastolic BP among runners immediately following a 4-h run at an
approximate speed of 6 miles per hour (63). PEH symptoms, such as dizziness,
blurred vision and syncope have been observed in individuals after
engaging in several forms of physical activity, such as walking,
jogging, cycling, swimming and resistance exercise (64,65). A
further explanation of the definitions of the different patterns of
BP in patients with PD is provided in Table I.
Heart rate is a basic indicator of autonomic
function, and the study of its measurement has been performed for a
long time in various traditions, even before the advent of modern
medicine (66). Additionally, it is
the sole indicator of autonomic function that can be readily and
accurately assessed, making it well-suited for extensive
population-based or epidemiological studies. A significant
association has been found between an increase in resting heart
rate over a period and an elevated risk of mortality from ischemic
heart disease and mortality from all causes (67). Increased mortality is linked to a
slower heart rate recovery following exercise (68). From a molecular standpoint,
administering high doses of β-adrenergic blocking medications only
results in a modest reduction (5-10 beats) in the resting heart
rate. On the other hand, the blockage of muscarinic receptors with
atropine leads to a significant rise (~40 beats) in the resting
heart rate (69). These discoveries
have led to the idea that the resting heart rate is mostly
controlled by vagal tone. The loss of this tone can have
pathological consequences, possibly as it usually helps to prevent
life-threatening irregular heart rhythms (70).
Heart rate variability (HRV), similar to heart rate,
can significantly correlate with morbidity and mortality (71). HRV assessed by time domain or
frequency domain techniques, is a non-invasive approach to evaluate
autonomic function by examining how neural mechanisms affect the
sinus node (72). HRV in the time
domain may exhibit normal patterns but decline in the mid and late
stages of PD (73). Power spectral
analysis of HRV can serve as a screening method to detect autonomic
dysfunctions in patients with PD by identifying low resting LF and
HF powers. PD can result in simultaneous impairment of both
sympathetic and parasympathetic nerves (74). Besides heart rate and HRV, another
key component in evaluating dysautonomia is the heart rate response
to deep breathing (75).
Head-up tilting is a standard and valuable test for
assessing cardiovascular autonomic function, particularly in
detecting OH. This can also be accomplished in the clinic by
transitioning from supine to seated or standing positions.
Measuring BP continuously during the exam is preferable, but
regular readings with an upper-arm sphygmomanometer can still be
sufficient (76). In addition,
several patients may be asymptomatic despite having OH, leading to
a lack of recognition by doctors.
The BP reaction to the Valsalva maneuver (VM), where
the pressure in the chest is increased to a maximum of 40 mmHg,
relies on the functioning of the baroreflex pathways. This reaction
is often abnormal in patients with autonomic failure and is
commonly employed to assess cardiovascular autonomic function.
Patients with PD with OH and up to 25% of patients with PD exhibit
abnormal baroreflex-cardiovagal gain responses, which are
determined during the VM (77). The
baroreflex sensitivity (BRS) VM approach has exhibited a stronger
association with cardiovascular autonomic function than the
spontaneous BRS indexes derived by the sequence or spectrum method.
BRS VM, as opposed to spontaneous BRS, has demonstrated a
prognostic significance in predicting the presence of
cardiovascular autonomic neuropathy according to the diagnostic
criteria established by the composite autonomic scoring scale in
patients with PD (78).
Ambulatory blood pressure monitoring (ABPM) is
valuable for identifying SH and PPH or OH, which can significantly
affect circadian BP patterns. PPH is most commonly observed
following the initial two meals of the day (79). Non-dipping, as previously mentioned,
is a lack of decrease in BP during the night, and it is frequently
observed in PD and autonomic dysfunction, resulting in a reversal
of the usual circadian BP pattern. Furthermore, 24-h BP recordings
offer clinicians valuable insight into the daily variations in BP,
aiding in the development of a treatment plan (including the timing
of medication administration and choice of medication) for
conditions such as PPH, OH and nocturnal SH (80).
The meal challenge test is used to identify the
presence of PPH. Typically, BP is monitored while the patient is
lying down and during fasting, both before and up to 120 min after
consuming a standardized meal. PPH is described similarly to OH,
characterized by a reduction in systolic BP of 20 mmHg after a meal
(58).
When assessing patients with PD, both while they are
taking medication and when they are not, it has been shown that
their BP and heart rate increase less during exercise compared to
individuals without PD. This phenomenon appears to be directly
linked to the disease itself and is not influenced by the drug used
to treat PD (81). Exercise testing
can detect exercise-induced or PEH in patients with autonomic
failure (63).
The association between plasma catecholamines,
particularly noradrenaline and outcomes in congestive heart failure
is a prominent example of this interaction (82). The association between elevated
noradrenaline levels and deteriorating outcomes in heart failure
supports the effective (but first contentious) utilization of
β-blocking medications in heart failure treatment (83). The primary constraint of plasma
noradrenaline (and adrenaline) in assessing autonomic function is
that it provides a momentary peek of activity unless multiple
measurements are taken throughout time. In addition, values
evaluated in the plasma indicate the overall equilibrium between
neural release, neural reuptake and various clearance methods. For
instance, under certain situations, such as severe hypoxia, there
may be noticeable enhancements in sympathetic neuronal activation
but only minimal increases in plasma noradrenaline (84). Patients diagnosed with PD who do not
have OH have been observed to exhibit minimal alterations in their
baseline supine resting and orthostatic plasma norepinephrine
levels. During the initial phase of PD without OH, there may be a
modest increase in plasma norepinephrine levels in certain people.
On the other hand, individuals with PD who experience OH may
exhibit lower-than-normal levels of BP both when standing and at
rest, indicating dysfunction of the autonomic nervous system
(39).
The skin vasomotor reflex is a term used to describe
the reduced cutaneous blood flow in the palm or sole provoked by
certain procedures, such as deep inspiration, mental stress, and
isometric exercise. Patients with Lewy body disease had a weakened
cutaneous vasomotor reflex (85).
The discoveries align with Lewy body disease findings in the raphe
nucleus, a region that significantly impacts the cutaneous
vasomotor reflex. Additionally, these findings may indicate the
involvement of postganglionic sympathetic pathways, which are
commonly affected in individuals with Lewy body disorders (86). The skin vasomotor reflex, which may
be evaluated with a Doppler flowmeter, frequently undergoes changes
in Lewy body illnesses following various adrenergic stimuli
(87).
Cardiac denervation occurs due to the loss of
postganglionic sympathetic neurons. These neurons release
catecholamine neurotransmitters, which bind to cardiac adrenergic
receptors (primarily B1 receptors). Catecholamine reuptake is a
normal function of postganglionic neurons. MIBG is an analog of
norepinephrine that utilizes the reuptake transporter system to
accumulate in postganglionic sympathetic neurons (88). Rissardo and Fornari Caprara (89) found that the early and delayed
registration heart-to-mediastinum ratios (H/M ratio) for diagnosing
PD were 1.70 and 1.51, respectively. In addition, the mean cutoff
for the early and delayed phases was 1.89 and 1.86(89).
Research has demonstrated reduced myocardial MIBG
uptake in patients with PD, indicating cardiac sympathetic
dysfunction (90). Notably, this
reduction is evident even in early-stage PD, suggesting that
myocardial MIBG scans could be valuable for the early detection of
PD. In this context, Kim et al (91) discovered that OH was closely
associated with cardiac sympathetic denervation observed on cardiac
MIBG in patients with early and mild PD. Furthermore, the decrease
in MIBG uptake is not exclusive to PD patients with symptomatic OH
or other dysautonomic symptoms (17,92).
Positron emission tomography (PET) scans using
6-[18F] fluoro-dopamine (18F-DA), another catecholamine analog,
have also shown decreased uptake in patients with PD (93). Tyrosine hydroxylase can also be used
to address the functionality of cardiac sympathetic postganglionic
neurons (94). MIBG and PET (6F-DA)
scans have shown that cardiac sympathetic denervation occurs
independently of OH. This suggests that damage to cardiac nerves
may precede damage to peripheral autonomic nerves. The latter's
dysfunction, leading to inadequate vasoconstriction, is considered
a significant factor in the development of OH (95).
Patients with autonomic dysfunction often encounter
hemodynamic abnormalities, such as nOH and SH, in addition to
genitourinary and gastrointestinal deficits. OH arises due to
sympathetic noradrenergic dysfunction and is clinically significant
in 20-50% of patients with PD (18,96,97).
nOH is defined as a decrease in systolic BP ≥20 mmHg
or a decrease ≥10 mmHg in diastolic pressure within 3 min after
transitioning from a supine to a standing position, without any
connection to cardiogenic causes or hypovolemia, which are
classified as non-nOH. It is advisable to extend testing in certain
patients, as they may experience delayed OH. In up to 50% of cases,
delayed OH can progress to classic OH after 10 years (33,98).
The decrease in BP during an orthostatic test is
influenced by factors, such as the duration of rest before
measuring supine BP, the method of assuming an upright position
(active standing or passive tilting) and the time spent standing.
In PD, OH occurs more frequently after tilting than during simple
standing and is often characterized by delayed onset (99).
Symptomatic OH manifests as lightheadedness,
presyncope, syncope, dizziness, visual disturbances, generalized
weakness and fatigue. Symptomatic and asymptomatic OH are both
linked to a higher incidence of hospital admissions, falls and a
reduced quality of life (100,101).
The findings presented in the study by Longardner et al
(102) support previous evidence on
the strong association between OH and cognitive impairment in PD
(31,103).
Various pathological mechanisms contribute to the
association between OH and cognition. These mechanisms include
cerebral hypoperfusion resulting from recurrent episodic
hypotension, widespread neurodegeneration, and dysfunction in
central and peripheral noradrenergic systems (103,104).
OH is considered one of the predictors of PDD in addition to age,
male sex and MCI (105).
Some authors have investigated the association
between OH and posture-related cognitive impairment in PD,
comparing cognitive function between patients with PD with and
without OH to healthy controls (107,108).
While in the supine position, both PD groups exhibited cognitive
deficits related to frontostriatal and visuospatial functions.
However, a transient improvement in cognition upon assuming an
upright-tilted position was observed only in the PD group with OH
(PD-OH). Furthermore, among patients with PDD, there was a greater
decrease in systolic BP and a more pronounced attention impairment
during standing (109).
The causative or associative nature of the
association between OH and cognitive dysfunction remains uncertain
(103). This uncertainty arises
partly as certain studies lack adjustments for critical variables,
such as comorbidities, medications and age. Further research is
required to elucidate this complex association.
nOH is closely linked to SH, which represents a
hemodynamically contrasting form of BP dysregulation. Approximately
half of the patients with PD-OH have concurrent OH and SH (110,111).
While expert consensus remains elusive regarding the
diagnostic criteria for SH, it is generally defined as having a
systolic BP ≥140 mmHg (or 150 mmHg) or a diastolic pressure ≥90
mmHg following a minimum of 5 min of rest while in a supine
position (112).
In addition, patients with SH frequently exhibit an
abnormal nocturnal BP profile (113). SH may be associated with either a
reduction in the typical nocturnal BP decline (usually ≥10%),
referred to as ‘non-dipping’, or an elevation in nighttime BP,
known as ‘nocturnal hypertension’ or ‘reverse dipping’ (114,115).
The underlying mechanisms of SH may arise from a
combination of baroreflex failure and vascular hypersensitivity. SH
is likely linked to less severe peripheral sympathetic denervation
than OH alone. Independent risk factors for SH include an older
age, the akinetic-rigid motor subtype (which predisposes to
cognitive decline), and pre-existing hypertension (116,117).
In PD, an irregular nocturnal BP profile often
indicates autonomic dysfunction. Normally, BP follows a circadian
rhythm, with a decrease >10% at night, known as ‘dipping’. The
majority of patients exhibit either non-dipping patterns or reverse
dipping patterns, known as ‘risers’. Non-dippers experience a loss
of nocturnal BP decline, while reverse dippers exhibit increased BP
values during the night (114,119).
In PD, disruptions in the circadian BP pattern have
been linked to coronary heart disease, stroke and higher mortality
rates. Additionally, these disturbances are associated with target
organ damage, cognitive impairment in older adults, and an
increased burden of white matter hyperintensities (WMHs) (120).
To the best of our knowledge, there are limited
studies available in the literature regarding nocturnal BP and
circadian rhythm alterations in patients with PD. Oh et al
(120) concluded that in patients
with PD, the presence of nocturnal hypertension was associated with
an elevated WMH score. Nighttime systolic pressure is closely
associated with white matter changes. Additionally, blunted HRV and
a lack of nocturnal decline in heart rate are related to increasing
WMHs scores. Of note, the non-dipping phenomenon does not appear to
influence WMHs. These findings suggest that white matter
alterations are linked to circadian autonomic dysfunction,
particularly nocturnal hypertension, in patients with PD.
Moreover, the presence of WMHs is significantly
associated with the risk of PDD (121). Consequently, circadian BP
disruptions may contribute to cognitive impairment in PD through
various distinct and independent mechanisms.
While cognitive impairment in PD may be linked to
cardiovascular dysautonomia, including BP dysregulation, the
association between these factors in dementia with Lewy bodies
(DLB) remains uncertain. Oka et al (122) aimed to investigate whether
cardiovascular dysautonomia affects cognitive function in Lewy body
disease. Their study evaluated 99 patients with de novo PD
(n=75) and DLB (n=24) using the mini-mental state examination
(MMSE) and frontal assessment battery (FAB). Additionally, they
estimated cardiac MIBG scintigraphy, assessed OH, SH and PPH,
analyzed nocturnal BP changes in 24-h ABPM and evaluated
constipation (122). The results of
their study were that in DLB, cardiac MIBG uptake was reduced, and
OH, PPH and SH were severely disturbed compared to PD.
Additionally, the nocturnal BP decline in 24-h ABPM was lower in
DLB. In PD, the failure of nocturnal decline was associated with
MMSE scores after adjusting for other clinical features.
Furthermore, the FAB was significantly associated with nocturnal BP
decline, age, and SH in PD, but no significant associations among
these factors were found for DLB (122).
The notable association between disrupted nocturnal
BP regulation and cognitive or executive decline in PD may be
attributed to impaired microvascular circulation or the
infiltration of α-synuclein in the central nervous system.
Conversely, the absence of an association between BP insufficiency
and cognitive impairment in DLB implies that Lewy body pathology
initially affects the neocortex, irrespective of autonomic nervous
system involvement (122). A
summary of the key differences between PD and DLB is presented in
Table II.
The cognition defect may occur before, after, or
even at the diagnosis time of PD in a variable degree of severity
(123). Previous studies have
proven that patients with PD have a much higher risk of developing
dementia than individuals without PD (124,125).
The risk of developing dementia among these patients reaches
~24-31% (126). Following the
diagnosis of PD by 10 years, approximately half the number of
patients have reported a risk for dementia and, in most cases, they
developed dementia after almost 20 years of diagnosis. Several risk
factors can lead to dementia in these patients. It was noted that
the risk of cognitive affection in PDD and AD is almost the same
(3). These patients require
assistance and may become dependent on others (127).
Recently, there has been increasing interest in the
stages that precede dementia in cognitive impairment, particularly
MCI (128). Studies have proven
that almost one-quarter of patients who have parkinsonism only
without dementia suffer from MCI (129). Other studies have proven that
almost 20% of patients suffered from MCI at the time of diagnosis
and this increased to ~50% in the fifth year of the disease
(130-132).
MCI is a transitional cognitive stage between normal and dementia
(133). The course of MCI is very
variable and may return to normal cognition in nearly one-quarter
of patients (133). However, even
after returning to normal, the case may deteriorate to dementia
again (134).
Studies have proven that patients with PD who have
memory issues at diagnosis and before having any treatment are at a
higher risk for developing MCI than those who do not initially have
memory issues (135,136). However, a number of factors can
affect the deterioration of MCI, particularly the affective
symptoms (137).
Males are more predominantly affected by cognitive
dysfunction than females. A previous cohort study demonstrated that
the majority of cognitive parameters, such as semantic fluency,
MoCA and phenomic parameters, apart from MMSE, are worse in males
which has exhibited no difference in both sex groups. In addition,
OH and RBD mostly affect males (138). Augustine et al (139) noted no difference in the age of
disease onset, motor symptoms, or diagnosis in each sex. However,
measures on symbol Digit and Scales for Outcomes of Parkinson's
Disease-Cognition (SCOPA-COG) were more improved in females
(139).
The association between cognitive function and male
sex remains unclear. OH and rapid eye movement (REM) have been
shown to be associated with lower cognitive function, and these
symptoms are more common in males. However, all these studies
(139,140,141)
included affected patients in the early stages of the disease;
thus, further research is required to determine whether this effect
will last in the late stages of the disease (143). Postmortem studies have revealed
that in PD, both cortical and limbic Lewy bodies are associated
with a higher risk of dementia. Higher levels of α-synuclein, along
with higher oligomeric forms, are associated with greater cognitive
impairment (144-146).
In addition, tau pathology and amyloid play a role in cognitive
dysfunction (147,148). Smith et al (149) recently reported that a large number
of patients with PD who have dementia had the same pathological
findings of AD in the form of moderate to severe pathology (tau and
amyloid-β). Moreover, the cerebrospinal fluid (CSF) of subjects
with PDD has been found to be associated with less amyloid-β than
affected patients without dementia or normal population. Low levels
of amyloid-β lead to the progression of dementia (150,151).
PD usually leads to sleep disorders, which may
affect cognitive function. Even in the early stages of PD with REM
disorders, patients are at a high risk of developing MCI. The risk
of developing MCI is higher among patients with REM at the baseline
(155). In addition, during the
early stages of the disease, mood changes may lead to further
cognitive dysfunction. However, the association between depression
and cognitive dysfunction remains unclear (156). Some studies have demonstrated a
strong connection between mood changes and cognition, although
others have not found such a connection (157).
Diseases such as hypercholesterolemia, hypertension,
obesity and diabetes are associated with dementia and cognitive
disorders (158). Furthermore, in
PD, these factors may worsen cognition. It has been found that body
mass index, hypertension and diabetes mellitus were associated with
hyperintensity of the brain white matter, which indicates ischemia
and predicts cognitive affection (159-161).
A previous cohort study revealed that high levels of C-reactive
protein, glycosylated hemoglobin and high levels of fasting blood
glucose during the early stages of PD are associated with low
scores on MMSE (162). Another
study proved that a high body mass index at the baseline led to a
more rapid decrease in cognitive function in the early stages of PD
(156). On the other hand, patients
who lose weight during the disease course can exhibit a more rapid
decline in cognitive function (163,164).
However, obese patients have a slower rate of cognition and
affection, particularly in memory and language (164). Thus, the association between body
weight and cognition affection appears to be very complex.
Changes in blood in the form of OH or SH that occur
in PD may increase the risk of developing dementia (165). In the case of OH, the repeated
hypoperfusion changes and changes in the cerebral blood flow may
cause cognitive affection (166).
SH can increase the risk of cerebral ischemia and, thus, cognitive
dysfunction, as aforementioned (159). Other research has reported a minor
effect of ischemic cerebrovascular lesions on PDD, to no relation
between the ischemic lesions in the heart and dementia (167). Diabetes mellitus is also associated
with a high risk of cognitive affection in PD. Studies have found
shared pathways between PD and diabetes mellitus (168-171).
Bosco et al (172) reported
that insulin resistance and abnormal metabolism of glucose were
more common among patients with PDD than among those who did not
have dementia.
Urate is a natural and crucial antioxidant in humans
and has an inverse effect on PD (173-175).
The uric acid levels in patients with PD are considered to be
associated with oxidation, chelation, genetics and apoptosis
(176). The uric acid level is low
in the CSF of patients with PD, whether they have dementia or not
(177). A low level of uric acid is
a poor prognostic factor for memory affection, attention and global
cognition (178-180).
Neuroinflammation inversely and strongly affects the
cognitive function in PD. The activation of microglial cells leads
to the release of cytokines, such as INF-γ, IL-1β, IL-6 and TNF-α,
damaging the dopaminergic neurons (181). in addition, the activation of
microglial cells decreases glucose metabolism in the frontal lobe
and other regions of the brain in patients with PD affected by
dementia (182). Mitochondrial
disorders increase oxidative stress pathways and astrocyte and
glial dysfunction, leading to abnormal inflammation (183-185).
There is a decrease in the activity of mitochondrial complex 1 and
low levels of mitochondrial DNA in the cortex of patients with PDD
(186).
Traumatic injury to the brain is a key factor for
disability. This disability may be due to inflammation, oxidative
stress, neuron death, blood-brain barrier breakdown, or brain edema
(187). PD is mostly associated
with frequent trauma (188).
Schiehser et al (189)
reported a greater decline in cognitive function among patients
with PD who had brain injury than who had not suffered any trauma.
The cognitive affection was mainly in the areas responsible for
memory and execution (189).
However, further studies are required to fully elucidate this
matter.
Some genes may affect the risk of developing PD and
contribute to PDD. One of these genes is apolipoprotein E (APOE)
(190). According to a previous
prospective study (191), this gene
inversely affects cognition. However, another study in the United
Kingdom found no association between this protein and cognitive
affection after almost 5 years (192). Thus, the effect of APOE on
cognition among patients with PD remains elusive.
Microtubule-associated protein tau plays a key role in microtubule
stabilization and assembly. The H1/H1 genotype of this protein
increases the risk of developing PD. The H1/H1 genotype leads to
the progression of dementia, although this effect decreases along
the disease course (193). This may
support the notion that the effect of this genotype on cognition
occurs only at the early stages of the disease (143).
Brain-derived neurotrophic factor (BDNF) is present
in a high amounts in the cortex and subcortex. One of its key
functions in the substantia nigra during development is to
establish the dopaminergic neurons (194). One of its variants is the G196A
(Val66Met) polymorphism (195). A
previous study demonstrated that in PD, patients who are carriers
of the BDNF Met gene are at a high risk of developing cognitive
disorders (196). However, other
studies have not found such a connection (197-199).
Thus, the effect of BDNF on cognition in PD remains unclear.
In Parkinsonism, neuropathology briefly consists of
the loss of dopamine neurons from the substantia nigra and the
abnormal deposition of α-synuclein, leading to the formation of
Lewy bodies. Firstly, this deposition occurs in the olfactory
system, monoaminergic and cholinergic neurons of the brainstem,
leading to synaptic affection (200). When the patient is affected by AD
and PD, the pathology is almost the same as previously described
(201), apart from deposition and
synaptic affection that occur in the limbic system rather than the
brainstem (201). The cognitive
defect that occurs in PD is considered to be due to
neurodegeneration that occurs in the brain rather than a functional
defect (202).
When MCI is associated with PD, there is more
degeneration in the dorsal striatum and the associative caudate
nucleus. These patients exhibit some preservation of dopamine
neurons in the brain (203).
However, dopamine neurons are markedly lost in PDD, particularly in
the temporal, parietal and frontal cortex (203). Normally, dopamine plays a crucial
role in maintaining cognition by reinforcing memory, attention,
cognitive effort and visuospatial functions (204,205).
Neurons that synthesize noradrenaline are present
in the locus coeruleus, which also produces neuromelanin in humans
(206). These neurons encourage
arousal and waking, and play a critical role in cognition,
particularly in long-term memory, attention, working and behavior
(207). The noradrenergic fibers
are arranged into two regions, the hippocampus and frontal cortex,
and are crucial for cognition (207). An association has been noted
between neuromelanin produced in the locus and MCI in PD cases
(207). In addition, if patients
have PD along with RBD, there is a similar and positive association
between neuromelanin reduction and cognition and OH (208,209).
Moreover, a deficiency in the transporter of noradrenaline in the
brain in PD is associated with OH and low levels of cognition
(209). If the patient has both PD
and OH (which is due to the cutting of noradrenaline supply to the
heart), the subject will suffer from cognitive dysfunction
(102). This may be due to cerebral
hypofunction resulting from OH, which leads to cognitive defects
(103,210). The DNA of noradrenaline neurons is
more liable to oxidative damage than others, which is a main
concern in patients with OH (211).
Noradrenaline markers are reduced to a high degree
in patients with both PD and dementia (212). Depending on the available data,
noradrenaline can be used as a parameter for cognitive affection in
various neuronal diseases, such as Parkinsonism (213). The density and volume of the basal
forebrain cholinergic area (the main source of acetylcholine
innervation to the amygdala, neocortex and hippocampus) are reduced
in PD in both newly diagnosed patients and during the course of the
disease (214-216).
This area is crucial for cognition, particularly memory, attention
and execution (217,218). In patients with dementia and PD,
the loss is mainly in cholinergic fibers, not neurons (219). The loss of acetylcholine
innervation to the cortex independently leads to a decreased
cognitive level. This is associated with dopamine loss and further
cognitive defects (215,220). Dopamine terminals heavily innervate
the cholinergic area of the forebrain, which may cause more
cognitive defects in these cases (221). In addition, the loss of cholinergic
innervation from the forebrain to the hippocampus leads to memory
troubles and deterioration to dementia (216,222).
In cases of PD with MCI, there is a decrease in acetylcholine
fibers in the hippocampus and their activity. Still, in cases
having both PD and dementia, there is also the deposition of
α-synuclein in the hippocampus and forebrain basally (223,224).
The mechanisms through which the cholinergic system in the basal
forebrain degenerates have not yet been fully elucidated. The
noradrenergic system in locus coeruleus is more exposed to
oxidative damage than cholinergic neurons (211). Following the decrease in
acetylcholine fibers in the cortex, α-synuclein is widely and
non-non-specifically aggregated in multiple neurons and
neurotransmitters of variable types (224). Fibers that contain galanin
increasingly innervate the cholinergic neurons in the forebrain,
basically in PD cases at the time of MCI development and
progression to dementia, which may be a cell response to injury
that occurs following α-synuclein aggregation (219).
Although serotonergic neurons are lost early from
the brainstem and even before the loss of dopaminergic neurons,
there is no definite association between the loss of serotonin
neurons and cognitive defects in PD (225). Serotonin loss is associated with
defects in motor and some non-motor functions, such as anxiety,
depression and sleep disorders (226,227).
The loss of serotonin from the brain in PD is likely due to
β-amyloid deposition, and drugs that increase serotonin
transmission decrease β-amyloid and cognitive defect (202). In PDD, α-synuclein deposition is
the cause of cognitive affection and other age-related pathologies
(228). Inflammation of the neurons
occurs only when Lewy pathology is associated with AD (229). In the majority of cases of PDD, the
limbic system, with or without the neocortex is affected by Lewy
pathology, but some cases do not have the same pathology (149). In mild cognitive impairment in
Parkinson's disease (PD-MCI), the entorhinal cortex is atrophied,
which affects memory (230).
α-synuclein deposition in this region leads to disease progression
to dementia (231). It has been
shown that α-synuclein deposition in the neocortex is the main
cause of PDD (149). Furthermore,
α-synuclein affects the DNA of the neuron and its repair (232). Of note, SNCA (the gene coding for
α-synuclein) is genetically variable and differs between PD and
DLB. Whether or not α-synuclein levels can predict cognitive defect
levels remains unclear (202).
Patients with PD and cognitive affection are likely
to have depositions of β-amyloid extracellular and tau
intracellular (aging pathology that occurs in AD) (147,150,233).
The deposition of β-amyloid precedes tau deposition and together
leads to AD (234).
The decreased activated glucocerebrosidase and low
potassium level increase α-synuclein phosphorylation and subsequent
cell pathology (240). A certain
polymorphism in the nucleotide of GBA decreases the expression of
glucocerebrosidase and increases α-synuclein deposition, leading to
PDD (241). APOE ε4 allele that
encodes APOE increases the cognitive defect (237). This allele may also increase the
deposition of β-amyloid in normal and diseased populations. Genetic
changes in SLC6A3 (or DAT that encodes for the transporter of
dopamine) are associated with poor cognition and reduced dopamine
transmission (242). There are
numerous genes that can affect cognition, such as those concerning
dopamine synthesis (DDC), degradation (COMT encodes for
catecholamine-O-methyltransferase) and receptors of dopamine
(DRD2, encodes for receptor 2 of dopamine) (243). Variations in the levels of these
genes may lead to cognitive disorders in PD (202).
The pathology of PD has a wide range of neuronal
affection that extends outside the dopaminergic nigrostriatal
system, leading to a number of non-motor issues. The accumulation
of Lewy particles in the neocortex and limbic system is considered
to be the cause of dementia and cognitive impairment in PD
(244). However, to date, studies
have not found a clear connection between the amount of Lewy
particles in the cortex and dementia in Parkinsonism (245). Similarly, Lewy particle
accumulation inside autonomic neurons peripherally and in
sympathetic ganglions may be associated with OH (autonomic
dysfunction) (246). Central
components of the autonomic system may also be involved,
particularly the hypothalamus, intermediolateral nucleus (in the
spinal cord), and vagus nucleus (dorsal part) (247). According to the study by Kosaka
(248), different disorders occur
due to Lewy molecules. The existence of these molecules and their
distribution in the brains of deceased individuals has helped
scientists to find certain criteria for each syndrome. Researchers
have found a variable connection between autonomic defects,
parkinsonism and dementia (249).
When Lewy bodies are present only in the brainstem, this leads to
idiopathic parkinsonism; however, when these bodies are present
widely in the cortex beside the brainstem, this leads to PDD. The
accumulation of these bodies in the sympathetic ganglion, spinal
cord and brain stem may lead to OH and parkinsonism (249) Previously, Braak et al
(250) extended this concept using
the stagewise hypothesis for synuclein progression in idiopathic
parkinsonism.
According to the available information, synuclein
is deposited extra-nigrally, firstly in the vagus nucleus
(dorsally) and the olfactory system, then nigral disorders in the
third stage, and finally in the cortex. This explanation does not
include the peripheral neurons (of the autonomic system) or the
spinal cord. However, it has been proposed that the efferent
sympathetic supply to the heart and intermediolateral nucleus are
the first sites to deposit Lewy particles in non-symptomatic
patients with PD (246). Thus,
perhaps the autonomic system plays a role in those having Lewy
particles in their cortex and complaining of dementia. Horimoto
et al (251) reported
autonomic issues in all deceased individuals who had Lewy particles
along with dementia in their study. These issues were variable from
OH, constipation and urinary incontinence. Orimo et al
(252) also reported the loss of
sympathetic innervation to the heart in all DLB cases in their
study In addition, OH has been noted to occur more among PDD
compared to those without dementia. Larner et al (253) also suggested that synuclein is
deposited firstly in the autonomic system and then in the cortex
after years.
Due to aging of the vascular and autonomic nerve
systems, as well as a decrease in baroreceptor sensitivity, OH is
common in the elderly (254).
Furthermore, any abrupt change in BP that causes a rapid and
considerable shift in cerebral blood flow may be considered to
produce or exacerbate cognitive impairment as aging is linked to
impaired cerebral autoregulation (255). The association between cognitive
impairment and OH in the elderly is presented in Table III. No marked differences were
reported in cognitive function and MMSE between individuals with
OH, and individuals without OH.
Autonomic dysregulation occurring in PD occurs by
both central and peripheral pathways (256). Aggregates of α-synuclein protein
forming Lewy bodies along with neuron loss are observed in the zona
compacta of the substantia nigra and the regions controlling the
autonomic functions (257).
Normally, baroreceptors maintain BP during standing
by releasing noradrenaline from the postganglionic neurons of the
sympathetic nervous system (104).
However, this mechanism is impaired in PD due to degeneration of
the postganglionic component of the sympathetic nervous system (the
main cause of dysautonomia affecting the heart in PD) (104,258).
In PD, the cutting of sympathetic innervation to the heart can be
shown using cardiac MIBG scintigraphy, which appears to have a low
myocardial uptake, and neuropathological imaging shows fiber loss
(259). Lewy bodies are
characteristic of neuropathology in PD and present in the cardiac
plexus of the affected individuals (260). In addition, sympathetic denervation
to the extracardiac blood vessels occurs, affecting both
noradrenaline release and vasoconstriction during standing
(258). Normally, the noradrenaline
level is doubled during standing after almost 5 min; however, this
level appears to be decreased in patients with PD-OH than in those
without OH (166,261). A progressive decrease in BP in
phase two of the VM in nOH, along with the loss of overshooting in
BP during phase four, suggests the loss of sympathetic baroreflex,
which may lead to systolic hypertension (258). nOH can result in a weak change in
the heart rate when changing positions from supine or standing. The
differentiation between nOH and non-nOH can be achieved by
monitoring the increase in heart rate and the fall in systolic BP.
nOH is associated with a mild increase in the heart rate and a gain
in baroreceptors by less than five beats per minute/mmHg (116,262).
In addition, Tipre and Goldstein (263) noted a decrease in the sympathetic
innervation to the kidney with blood volume depletion due to
diuresis and natriuresis. The management of PD mainly involves the
use of levodopa alone or along with benserazide or carbidopa,
leading to an increase in dopamine levels and its metabolites in
the plasma. This leads to vasodilation along with an increase in
natriuresis and diuresis, leading to a reduction of both
extracellular fluid and blood volume (258). The impairment of the sympathetic
innervation to the heart and baroreflex in PD leads to hypotension
(104). Furthermore, Noack et
al (264) presumed that the
negative inotropic effect that occurred with levodopa was the cause
of hypotension, not vasodilatation. In PD, the relation between
manifestations of dysautonomia and involved structures of the
autonomic nervous system remains unclear (256). The central biological clock and the
hypothalamus regulate the circadian rhythm of heart rate and BP
through the suprachiasmatic nucleus (SCN) (265). This nucleus activates the autonomic
innervation through both GABA neurons and the regulation of the
release of melatonin (265). A
previous study found that α-synuclein is deposited in the SCN of
affected individuals (257).
However, another study found a low serum melatonin level in the
affected people (266). In the
brain, noradrenaline is mainly produced by the nucleus coeruleus,
which is early affected in PD (267) along with a decrease in the
transporter of noradrenaline (209).
A reduced number of catecholamine neurons in the
solitary tract may cause impaired baroreflex (268). In patients with combined PD and OH,
an increased level of Lewy bodies in the cerebral cortex (insular
part) is noted with a defect in the functional connection between
the striatum, thalamus and hypothalamus (269,270).
WMHs are regions of the heightened signal detected
on T2-weighted or fluid-attenuated inversion recovery (FLAIR)
magnetic resonance images. These hyperintensities primarily arise
from long-term, widespread and asymptomatic ischemia (i.e., reduced
blood flow to brain tissue, leading to insufficient oxygen and
glucose supply and disrupting cellular metabolism). While
periventricular regions are primarily affected, WMHs can occur
throughout the entire brain (271,272).
WMHs are commonly observed in elderly individuals, as well as in
individuals diagnosed with AD. These WMHs are indicative of the
existence of demyelination and axonal degradation (272). WMHs can have a significant effect
on the cognitive function of both healthy older patients, and
patients with MCI and dementia (273,274).
In PD, WMHs have been linked to OH (35,120).
The association between the WMHs and OH supports the theory that
repeated episodes of hypotension may result in reduced blood flow
to the brain, potentially causing damage to susceptible brain
regions and cognitive impairments (103). Moreover, the degenerative
mechanisms associated with age and PD may contribute to the changes
in white matter, possibly due to vascular insufficiency (120).
Early WMHs have also been associated with cognitive
impairments and a subsequent higher likelihood of developing
dementia in individuals with PD (275,276).
Examining the temporal linkages between dysautonomia, WMHs and
cognitive decline could provide insight into the fundamental
mechanisms of these significant non-motor symptoms in PD.
Furthermore, studies have revealed that in patients
with PD and other synucleinopathies, such as multiple system
atrophy, dementia with Lewy bodies and pure autonomic failure, SH
plays a crucial role. Specifically, the average BP measurement in a
supine position was the most reliable indicator of target organ
damage. Additionally, SH was independently associated with a
greater burden of WMHs, along with more severe renal failure and a
higher prevalence of left ventricular hypertrophy (118). Similarly, within the overall
population, experiencing reverse dipping (nocturnal hypertension)
is significantly linked to an increased risk of developing cerebral
small vessel disease and cognitive impairment (277). Cerebral small vessel disease is the
primary cause of vascular dementia, encompassing lacunar infarcts
(LCI) and WMHs (278). Hypertension
significantly increases the likelihood of experiencing cognitive
impairment, and there is a clear association between reduced brain
blood volume, neuritic plaques and hypertension (279). Earlier onset and more severe
manifestations of OH are associated with an increased risk of
developing dementia in patients diagnosed with PD. Some authors
have hypothesized that individuals with earlier and more severe OH
will exhibit more severe brain disorders characterized by the
accumulation of abnormal proteins and/or abnormalities in the blood
vessels, particularly in the presence of concomitant SH (280). Due to the simultaneous occurrence
of OH and SH in the same patient, separating and understanding
their individual implications is challenging. OH has both
immediate, brief effects, such as fainting and accidents, as well
as long-term issues such as kidney failure (41). The difficulty in distinguishing the
effects of OH from those of SH leads to conflicting opinions among
clinicians regarding the optimal approach to managing both
conditions, as addressing one condition typically worsens the other
(31,104). A schematic summary of the
pathophysiology of autonomic dysfunction and cognitive impairment
in PD is presented in Fig. 2.
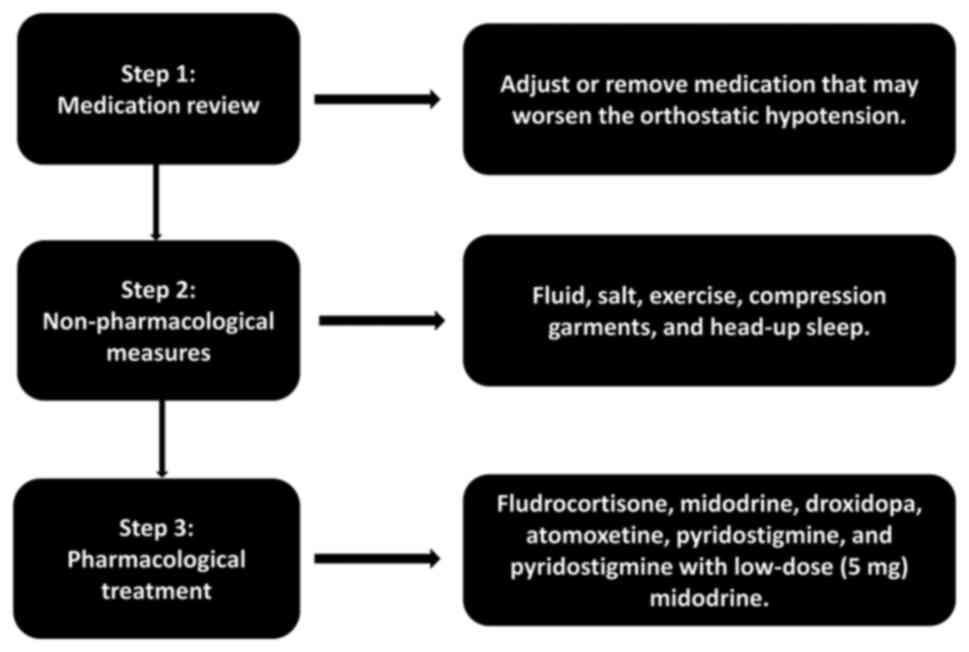
The primary goal of treating patients with nOH with
synucleinopathies is not to achieve a normal standing BP, but
rather to decrease the severity of symptoms, enhance the quality of
life, and diminish the risk of complications and mortality
(281). There are established
criteria for the management of nOH. If nOH does not exhibit any
symptoms, treatment may not be necessary or may be confined to
non-pharmacological approaches. Pharmacological treatment is
typically necessary when nOH is symptomatic, causing symptoms of
organ hypoperfusion, such as dizziness, lightheadedness and blurred
vision (32,262). The management flowchart of OH is
illustrated in Fig. 3.
Syncope is often caused by straining and
Valsalva-like maneuvers during bowel movements (289). In this situation, the treatment of
constipation is necessary (290).
Compression garments can prevent syncope and should be considered
(291,292). Sleeping with the head up also has a
potential benefit in the management of nOH (291).
Fludrocortisone, also known as 9α-fludrocortisone,
is an artificial mineralocorticoid that raises BP through at least
one of the following pathways: It promotes sodium and water
reabsorption in the kidneys, increasing fluid volume within blood
vessels. Additionally, it improves the ability of the body to
respond to natural stress hormones and medicines that increase BP
(294). However, fludrocortisone
worsens sodium and water retention and contributes to the
progression of left ventricular hypertrophy and renal failure,
potentially increasing the likelihood of hospitalization for any
reason (295). Other common
side-effects include hypokalemia and ankle edema (294,296).
Fludrocortisone is usually prescribed at a daily dosage of 0.1-0.2
mg, and no significant improvement has been reported by increasing
the dose to >0.3 mg per day. In fact, increasing the dose beyond
0.3 mg per day is associated with an elevated risk of side-effects
(297-300).
Another key pharmacological class for the
management of nOH is the pressor agents. Midodrine is a type of
medication that is converted into a substance known as
desglymidodrine in the body. Desglymidodrine acts as a stimulant
for a specific type of receptor termed α1-adrenoreceptor, which
helps increase blood vessel resistance and BP. The recommended dose
ranges from 2.5 to 15 mg, taken once to thrice daily during waking
hours. For instance, a three-times daily plan could involve taking
the medication before bed, lunch and mid-afternoon (301). The administration of midodrine
leads to a notable elevation in both systolic and diastolic BP,
accompanied by slight enhancements in orthostatic symptoms.
Midodrine poses a notable risk of causing SH; thus, it is advised
that patients refrain from taking midodrine within 5 h of going to
bed (28,302,303).
Stimulating α1-adrenergic receptors can lead to undesirable
effects, including piloerection (often known as ‘goosebumps’),
itching of the scalp and urine retention. Midodrine does not affect
the heart rate, since it does not stimulate β-adrenoreceptors.
Additionally, it has no deleterious effects on the central nervous
system due to its limited ability to penetrate the blood-brain
barrier (304).
Droxidopa, also known as
L-threo-3,4-dihydroxyphenyl-serine or L-DOPS, is an orally
administered synthetic amino acid that undergoes conversion to
norepinephrine within the body (305). The maximum levels of droxidopa in
the bloodstream are achieved within ~3 h following its oral
administration. The amount administered in clinical studies ranged
from 100 to 600 mg, taken three times per day. However, clinical
observations suggest that the dosage should be individualized based
on the specific needs of each patient, taking into account their
periods of activity and inactivity (305-307).
Due to the variability in the pressure impact of droxidopa across
patients, it is strongly advised to undergo a titration procedure
under the supervision of a clinician (281). Additional research is required to
determine whether droxidopa has positive effects on both motor and
non-motor symptoms caused by a lack of norepinephrine in patients
with PD (267). The FDA has
approved atomoxetine, a medication that selectively blocks the
norepinephrine transporter, for the treatment of attention deficit
hyperactivity disorder (308).
Prior research has demonstrated that administering a daily dosage
of 18 mg of atomoxetine effectively alleviates OH and symptoms
associated with nOH in patients (309,310).
Following atomoxetine delivery, elevated norepinephrine levels have
been linked to an elevated standing BP (311). Autonomic dysfunction is also
observed in patients with drug-induced parkinsonism, and
atomoxetine has shown promise in treating drug-induced
parkinsonism, particularly in patients whose elevated sympathetic
tone is a symptom of a psychiatric disorder (30,312)
In patients with nOH, either pyridostigmine alone
or combined with low-dose (5 mg) midodrine hydrochloride will
improve orthostatic BP without aggravating SH (313). However, the combination of 5 mg
midodrine and 60 mg pyridostigmine has exhibited better results
than either agent used alone. Pyridostigmine is less effective than
midodrine in alleviating symptoms linked to nOH (314). Case reports and proof of concept
studies have demonstrated the effectiveness of yohimbine,
ergotamine, dihydroergotamine, ephedrine, desmopressin,
indomethacin and fluoxetine in the treatment of nOH (315).
Various non-pharmacological approaches can be
employed to decrease nOH. Information about these methods should be
included in the education of patient on nOH (262,316).
Supine BP can be reduced by raising the head of the bed (111,317).
Patients should raise their heads off the beds 6 to 9 inches (~30˚)
when sleeping at night. Sleeping with the head up lowers BP and
natriuresis, while maintaining an activated renin-angiotensin
system, resulting in a less abrupt drop in BP upon waking (299). Another strategy for lowering
nocturnal hypertension is to consume a snack high in carbohydrates
before going to bed. This has two effects: First, it reduces BP by
transferring blood to the splanchnic circulation; second, it
triggers insulin release, which has a direct vasodilator effect
(305,318). Reducing BP can also be achieved
with the aid of local passive heat. Patients suffering from SH have
had their BP reduced overnight by using a heating pad set at
40-42˚C for 2 h (319). Using
continuous positive airway pressure (8-12 cm H2O)
overnight is a novel non-pharmacological method for treating SH.
This method reduced BP throughout the night, reduced diuresis
during the night, and improved morning symptoms of nOH (320).
A short-acting antihypertensive medication taken
before bed may be required if nSH continues to be present after
nonpharmacologic methods have been exhausted (321,322).
In general, patients with severe nSH (systolic/diastolic BP
≥180/≥110 mmHg) may require the pharmacological management of nSH.
By contrast, those with moderate nSH (BP 160-179/100-109 mmHg) may
be considered for pharmacological care, depending on their specific
case and risk profile (116,262).
When possible, it is best to use short-acting medications to treat
hypertension so that the symptoms of patients with nOH do not
worsen upon waking in the morning (322). Medications that can be used to
treat SH include captopril, eplerenone, hydralazine, losartan,
nifedipine and nitroglycerin patches, as summarized in Table IV.
The factors that can worsen cognitive impairment
need to be assessed and resolved. Polypharmacy, mental issues and
sleep disturbances are all examples of such conditions (323,324).
When feasible, it is best to avoid psychoactive substances.
Medications with minimal negative effects on cognition should be
evaluated when treatment is required.
As regards treating depression in PD, for instance,
paroxetine is effective. However, patients with PD who also have
cognitive impairment should not take paroxetine, since it has the
worst anticholinergic profile of all the depression drugs (325,326).
At each visit, it is critical to check for medications with direct
and indirect anticholinergic effects. Patients with PD should not
be initially prescribed the anticholinergic medicine oxybutynin for
the treatment of overactive bladder as it has been demonstrated to
reach the central nervous system and induce cognitive impairment in
certain individuals (327). It is
also critical to check for non-prescription drugs that can cause
side-effects, such as diphenhydramine-containing over-the-counter
sleep aids (328). Patients should
be advised to stop using these products and instead try sleep
hygiene techniques that do not involve medication or sleep aids
that target both sleep and any other symptoms that may be present,
such as depression, psychosis or hallucinations (329). Cognitive-based therapies are
distinct from non-pharmacological interventions, since they aim to
improve cognition rather than other behavioral or functional
objectives, including cognitive stimulation, training, and
rehabilitation (330). Cognitive
stimulation provides broader stimulation to enhance cognitive and
social performance. Cognitive training employs standardized
cognitive tasks administered through computer-based or paper-based
methods. Cognitive rehabilitation focuses on addressing specific
challenges in everyday tasks to enhance overall functioning
(330). Various physical workouts,
such as treadmill training, dance, stationary cycling, Wii Fit and
Tai Chi, have been assessed for their impact on cognition. An
extensive analysis of randomized controlled trials (RCTs) examining
the impact of physical exercise on cognition in individuals with
PD, including those with normal cognition and PD-MCI, revealed that
physical exercise resulted in enhancements in overall cognitive
function, processing speed, attention and mental flexibility.
Utilizing the treadmill three times per week for 60 min was shown
to result in the most significant improvement in cognition
(331). For those with mild to
severe PD with a MMSE score >24, a regimen of treadmill walking
consisting of 45 min per session, 3 days per week, for a duration
of 3 weeks was shown to lead to a substantial enhancement in
executive function as assessed by the FAB, trail-making and memory
interference tests (332).
Transcranial magnetic stimulation has been studied
in the context of PD for the treatment of motor, emotional and
cognitive symptoms. There are insufficient conclusive data to
support the effectiveness of repetitive transcranial magnetic
stimulation in enhancing cognitive function in individuals with
cognitive impairment associated with PD. A previous study
demonstrated that repetitive intermittent stimulation, known as
‘theta burst’ of the left dorsolateral prefrontal cortex (DLPFC)
resulted in enhanced cognitive abilities and visuospatial ability
that persisted for up to 1 month in individuals with PD-MCI
(333). Transcranial direct current
stimulation (tDCS) applied to the prefrontal cortex has been shown
to result in enhanced executive function, namely in trail-making
activities. Still, it has not led to similar improvements in other
tests, such as the Stroop test or the Wisconsin Card Sorting Test,
in patients with PD who did not have dementia (334). An RCT with a sample size of 22
participants demonstrated that the combination of cognitive
training and tDCS applied to the left DLPFC in adults with PD-MCI
resulted in improved phonemic verbal fluency compared to cognitive
training alone. This improvement was shown following a treatment
period of 5 days per week for 2 weeks and was sustained at the
3-month follow-up assessment. Nevertheless, there were no
discernible variations between the groups regarding other key
variables related to language, attention, and executive processes
(335).
The precise mechanisms responsible for the
cognitive decline in PD remain incompletely known. It is widely
recognized that patients with PD experience the early degeneration
of cholinergic neurons in the basal forebrain. These neurons supply
cholinergic connections to the neocortex (247,336).
As a result, the use of acetylcholinesterase inhibitors (AChEI) has
been thoroughly examined for the treatment of PDD. Rivastigmine is
an AChEI, one of the three AChEIs approved by the FDA for the
treatment of cognitive symptoms in AD. It possesses a distinct
characteristic of inhibiting acetylcholinesterase and
butyrylcholinesterase, which has been proposed as a possible
explanation for its effectiveness (337). The efficacy of rivastigmine was
established in a 24-week clinical trial with 541 individuals with
mild to moderate PDD. The trial was conducted double-blind with a
placebo control group (3). The doses
of the study medicine were gradually increased over 16 weeks, with
each patient being maintained on the greatest dose they could
tolerate for the remainder of the study. The average dosage of
rivastigmine reached 8.6 mg per day by the conclusion of the
dose-escalation phase. The main measures of effectiveness were the
scores for the Alzheimer Disease Assessment Scale-Cognitive
Subscale (ADAS-cog) and the Alzheimer's Disease Cooperative
Study-Clinician's Global Impression of Change (ADCS-CGIC). The
secondary outcomes included the Alzheimer's Disease Cooperative
Study-Activities of Daily Living (ADCS-ADL), the 10-item
neuropsychiatric inventory, the MMSE, the cognitive drug research
computerized assessment system, the Delis-Kaplan Executive Function
System (D-KEFS) Verbal Fluency Test, and the 10-point Clock Drawing
Test. The rivastigmine group demonstrated a mean improvement of 2.1
points on the ADAS-cog after 24 weeks of treatment, while the
placebo group experienced a decrease of 0.7 points. There was a
19.8% clinical improvement on the ADCS-CGIC in the group treated
with rivastigmine, compared to a 14.5% improvement in the group
treated with placebo (P=0.007). In addition, a significant increase
in clinically significant deterioration on the ADCS-CGIC scale was
detected in 13% of the participants in the rivastigmine group and
23.1% in the placebo group (P=0.007). Furthermore, rivastigmine
demonstrated a substantial advantage over placebo in all additional
measures of effectiveness. It is worth mentioning that a somewhat
higher number of patients in the rivastigmine group reported
experiencing symptoms such as nausea, vomiting, tremors, and
dizziness (3). Rivastigmine has the
potential to enhance apathy in PD, in addition to its effects on
cognitive impairment. A 6-month clinical experiment was conducted
to evaluate the effects of a rivastigmine transdermal patch on
apathy in 30 patients with PD who did not have dementia or
depression. The trial was double-blind, placebo-controlled, and
randomized. The results showed that the rivastigmine transdermal
patch substantially impacted the Lille Apathy Rating Scale (LARS)
in patients with moderate to severe apathy (338). Other treatment options, including
donepezil, galantamine, and memantine are summarized in Table V.
The current guidelines for the management of
arterial hypertension provide a comprehensive approach to treating
patients with comorbid conditions such as PD (44). Studies on cholinesterase inhibitors
and memantine have shown varied efficacy in improving cognitive
impairment in patients with PDD and dementia with Lewy bodies
(339-341).
Furthermore, treatment with cholinesterase inhibitors like
donepezil has demonstrated efficacy in clinical trials,
specifically in patients with PD dementia (342). Research also indicates that certain
interventions targeting orthostatic hypotension and syncope in
Parkinson's disease can have positive effects on overall cognitive
and functional outcomes (343-347).
In addition, the treatment of autonomic
dysfunctions with pharmacological agents, such as midodrine and
droxidopa has been evaluated, with findings supporting their use in
managing orthostatic hypotension and its associated cognitive
decline (348-353).
In recent years, notable achievements have been
made, such as the commencement and successful conclusion of
extensive clinical trials and the authorization of novel treatments
for autonomic dysfunction and cognitive impairment, each one
separately. However, investigating the association between
cardiovascular dysautonomia and cognition in PD can provide a
clearer understanding of the underlying mechanisms that connect
these significant nonmotor symptoms. This includes examining the
pathological and neurochemical connections.
Cognitive impairment may be directly and indirectly
related to autonomic dysfunctions. Further studies about the
pathophysiology of cognitive function in individuals with PD are
warranted, particularly as it affects 4 out of every 5 individuals
with PD. The current literature shows an indirect association in
which patients with autonomic dysfunctions have been observed to
have white matter changes (280).
It is worth investigating the cardiac function with cardiac MIBG
before developing peripheral symptoms and then evaluating
neuroimaging with different sequences and volumetric studies. It is
possible that the occurrence of autonomic symptoms is directly
associated with the progression of the disease, and at the same
stage, the patient develops independent cognitive dysfunction.
Notably, some studies have revealed improved cognitive function
with the optimized management of cardiovascular dysfunction
(89-91).
Conducting larger prospective studies that analyze
clinical, genetic, and pathological data will provide a better
understanding of the variability in autonomic and cognitive
impairment in PD. Moreover, it is necessary to conduct clinical
trials to examine whether dealing with dysautonomia during the
prodromal or early stages will decelerate or delay cognitive
deterioration. Additional information about the link between
dysautonomia and cognitive impairment may be revealed by functional
imaging, which might be combined with new magnetic resonance
imaging methods to assess connectivity. Understanding the rate and
pattern of cognitive decline becomes essential for future research,
as it varies significantly. Investigating the underlying mechanisms
behind various clinical patterns is an important priority.
In summary, the present review described a
significant association between cardiovascular dysautonomia, in
particular OH, and cognitive impairment. Dysautonomia, WMHs and
cognitive decline are major non-motor symptoms in PD, and
understanding their temporal and causal linkages may shed light
onto some of the underlying pathophysiological mechanisms. It is a
multifactorial process, and the most commonly suggested mechanisms
are hypoperfusion in the white matter of the brain or the spread of
α-synuclein protein from the peripheral autonomic nervous system to
the central nervous system, recurrent episodic brain
hypoxia/ischemia, which is linked to widespread neuronal apoptosis
and significant infarction. These findings imply that repeated
episodes of reduced blood flow and oxygen supply to the brain
caused by dysautonomia may lead to a higher likelihood of
significant accumulation of α-synuclein protein and, therefore, be
linked to cognitive loss. Thus, it is a vicious cycle and linked
mechanisms that finally lead to the development of this
association. If dysautonomia is identified as a potential
modifiable risk factor for cognitive impairment, then early
diagnosis and treatment would be an essential approach for
minimizing the risk of future cognitive decline. Hence, treating BP
dysregulation may impede the advancement of cognitive deterioration
in PD. Further clinical trials with a larger population are
required to confirm these findings on the pathogenesis of cognitive
impairment in the very early stages of PD, which may be
prevented.
Not applicable.
Funding: No funding was received.
Not applicable.
IK, RS, AMK, HHS, ALFC and JPR designed the study
and drafted the manuscript. IK, RS and JPR wrote the whole
manuscript. All authors have read and approved the final version of
the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Aarsland D, Marsh L and Schrag A:
Neuropsychiatric symptoms in Parkinson's disease. Mov Disord.
24:2175–2186. 2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hely MA, Reid WG, Adena MA, Halliday GM
and Morris JG: The Sydney multicenter study of Parkinson's disease:
The inevitability of dementia at 20 years. Mov Disord. 23:837–844.
2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Emre M, Aarsland D, Albanese A, Byrne EJ,
Deuschl G, De Deyn PP, Durif F, Kulisevsky J, van Laar T, Lees A,
et al: Rivastigmine for dementia associated with Parkinson's
disease. N Engl J Med. 351:2509–2518. 2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kehagia AA, Barker RA and Robbins TW:
Cognitive impairment in Parkinson's disease: The dual syndrome
hypothesis. Neurodegener Dis. 11:79–92. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mattay VS, Tessitore A, Callicott JH,
Bertolino A, Goldberg TE, Chase TN, Hyde TM and Weinberger DR:
Dopaminergic modulation of cortical function in patients with
Parkinson's disease. Ann Neurol. 51:156–164. 2002.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Olanow CW, Schapira AH and Roth T: Waking
up to sleep episodes in Parkinson's disease. Mov Disord.
15:212–215. 2000.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Louter M, Munneke M, Bloem BR and Overeem
S: Nocturnal hypokinesia and sleep quality in Parkinson's disease.
J Am Geriatr Soc. 60:1104–1108. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pfeiffer RF: Autonomic Dysfunction in
Parkinson's disease. Neurotherapeutics. 17:1464–1479.
2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Zhou Z, Zhou X, Zhou X, Xiang Y, Zhu L,
Qin L, Qin L, Wang Y, Pan H, Zhao Y, et al: Characteristics of
Autonomic Dysfunction in Parkinson's disease: A large chinese
multicenter cohort study. Front Aging Neurosci.
13(761044)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Blackett H, Walker R and Wood B: Urinary
dysfunction in Parkinson's disease: A review. Park Relat Disord.
15:81–87. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sakakibara R, Tateno F, Kishi M, Tsuyuzaki
Y, Uchiyama T and Yamamoto T: Pathophysiology of bladder
dysfunction in Parkinson's disease. Neurobiol Dis. 46:565–571.
2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sakakibara R, Uchiyama T, Yamanishi T,
Shirai K and Hattori T: Bladder and bowel dysfunction in
Parkinson's disease. J Neural Transm (Vienna). 115:443–460.
2008.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sakakibara R, Shinotoh H, Uchiyama T,
Yoshiyama M, Hattori T and Yamanishi T: SPECT imaging of the
dopamine transporter with [(123)I]-beta-CIT reveals marked decline
of nigrostriatal dopaminergic function in Parkinson's disease with
urinary dysfunction. J Neurol Sci. 187:55–59. 2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Winge K, Friberg L, Werdelin L, Nielsen KK
and Stimpel H: Relationship between nigrostriatal dopaminergic
degeneration, urinary symptoms, and bladder control in Parkinson's
disease. Eur J Neurol. 12:842–850. 2005.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fasano A, Visanji NP, Liu LW, Lang AE and
Pfeiffer RF: Gastrointestinal dysfunction in Parkinson's disease.
Lancet Neurol. 14:625–639. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Abbott RD, Petrovitch H, White LR, Masaki
KH, Tanner CM, Curb JD, Grandinetti A, Blanchette PL, Popper JS and
Ross GW: Frequency of bowel movements and the future risk of
Parkinson's disease. Neurology. 57:456–462. 2001.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Goldstein DS, Holmes C, Li ST, Bruce S,
Metman LV and Cannon RO III: Cardiac sympathetic denervation in
Parkinson disease. Ann Intern Med. 133:338–347. 2000.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Goldstein DS: Orthostatic hypotension as
an early finding in Parkinson's disease. Clin Auton Res. 16:46–54.
2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Fanciulli A, Strano S, Colosimo C,
Caltagirone C, Spalletta G and Pontieri FE: The potential
prognostic role of cardiovascular autonomic failure in
α-synucleinopathies. Eur J Neurol. 20:231–235. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Snider SR, Fahn S, Isgreen WP and Cote LJ:
Primary sensory symptoms in parkinsonism. Neurology. 26:423–429.
1976.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Goetz CG, Tanner CM, Levy M, Wilson RS and
Garron DC: Pain in Parkinson's disease. Mov Disord. 1:45–49.
1986.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Waseem S and Gwinn-Hardy K: Pain in
Parkinson's disease. Common yet seldom recognized symptom is
treatable. Postgrad Med. 110:33–34. 2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Nagy A, Eordegh G, Paroczy Z, Markus Z and
Benedek G: Multisensory integration in the basal ganglia. Eur J
Neurosci. 24:917–924. 2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Takakusaki K, Oohinata-Sugimoto J, Saitoh
K and Habaguchi T: Role of basal ganglia-brainstem systems in the
control of postural muscle tone and locomotion. Prog Brain Res.
143:231–237. 2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
von Coelln R and Shulman LM: Clinical
subtypes and genetic heterogeneity: Of lumping and splitting in
Parkinson disease. Curr Opin Neurol. 29:727–734. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Braak H, Ghebremedhin E, Rub U, Bratzke H
and Del Tredici K: Stages in the development of Parkinson's
disease-related pathology. Cell Tissue Res. 318:121–134.
2004.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chen Z, Li G and Liu J: Autonomic
dysfunction in Parkinson's disease: Implications for
pathophysiology, diagnosis, and treatment. Neurobiol Dis.
134(104700)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Jankovic J, Gilden JL, Hiner BC, Kaufmann
H, Brown DC, Coghlan CH, Rubin M and Fouad-Tarazi FM: Neurogenic
orthostatic hypotension: A double-blind, placebo-controlled study
with midodrine. Am J Med. 95:38–48. 1993.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kehagia AA, Barker RA and Robbins TW:
Neuropsychological and clinical heterogeneity of cognitive
impairment and dementia in patients with Parkinson's disease.
Lancet Neurol. 9:1200–1213. 2010.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rissardo JP and Caprara ALF: Risk factors
for Parkinson's disease dementia. Ann Mov Disord. 6:156–159.
2023.
|
|
31
|
Udow SJ, Robertson AD, MacIntosh BJ, Espay
AJ, Rowe JB, Lang AE and Masellis M: Under pressure. Is There a
Link between Orthostatic hypotension and cognitive impairment in
α-Synucleinopathies. J Neurol Neurosurg Psychiatry. 87:1311–1321.
2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Lahrmann H, Cortelli P, Hilz M, Mathias
CJ, Struhal W and Tassinari M: EFNS guidelines on the diagnosis and
management of Orthostatic hypotension. Eur J Neurol. 13:930–936.
2006.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Freeman R, Wieling W, Axelrod FB, Benditt
DG, Benarroch E, Biaggioni I, Cheshire WP, Chelimsky T, Cortelli P,
Gibbons CH, et al: Consensus statement on the definition of
Orthostatic hypotension, neurally mediated syncope and the postural
tachycardia syndrome. Clin Auton Res. 21:69–72. 2011.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rascol O, Perez-Lloret S, Damier P, Delval
A, Derkinderen P, Destee A, Meissner WG, Tison F and Negre-Pages L:
Falls in ambulatory non-demented patients with Parkinson's disease.
J Neural Transm(Vienna). 122:1447–1455. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Verbaan D, Marinus J, Visser M, van Rooden
SM, Stiggelbout AM, Middelkoop HA and van Hilten JJ: Cognitive
impairment in Parkinson's disease. J Neurol Neurosurg Psychiatry.
78:1182–1187. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Martinez-Martin P, Schapira AH, Stocchi F,
Sethi K, Odin P, MacPhee G, Brown RG, Naidu Y, Clayton L, Abe K, et
al: Prevalence of nonmotor symptoms in Parkinson's disease in an
international setting; study using nonmotor symptoms questionnaire
in 545 patients. Mov Disord. 22:1623–1629. 2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Cutsforth-Gregory JK and Low PA:
Neurogenic Orthostatic Hypotension in Parkinson Disease: A Primer.
Neurol Ther. 8:307–324. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Pitton Rissardo J and Fornari Caprara AL:
Parkinson's disease rating scales: A literature review. Ann Mov
Disord. 3:3–22. 2020.
|
|
39
|
Goldstein DS, Pechnik S, Holmes C, Eldadah
B and Sharabi Y: Association between supine hypertension and
Orthostatic Hypotension in autonomic failure. Hypertension.
42:136–142. 2003.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Vagaonescu TD, Saadia D, Tuhrim S,
Phillips RA and Kaufmann H: Hypertensive cardiovascular damage in
patients with primary autonomic failure. Lancet. 355:725–726.
2000.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Garland EM, Gamboa A, Okamoto L, Raj SR,
Black BK, Davis TL, Biaggioni I and Robertson D: Renal impairment
of pure autonomic failure. Hypertension. 54:1057–1061.
2009.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Minhas JS, Wang X, Lavados PM, Moullaali
TJ, Arima H, Billot L, Hackett ML, Olavarria VV, Middleton S,
Pontes-Neto O, et al: Blood pressure variability and outcome in
acute ischemic and hemorrhagic stroke: A post hoc analysis of the
HeadPoST study. J Hum Hypertens. 33:411–418. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pickering TG, Hall JE, Appel LJ, Falkner
BE, Graves J, Hill MN, Jones DW, Kurtz T, Sheps SG and Roccella EJ:
Subcommittee of Professional and Public Education of the American
Heart Association Council on High Blood Pressure Research.
Recommendations for blood pressure measurement in humans and
experimental animals: Part 1: blood pressure measurement in humans:
a statement for professionals from the Subcommittee of Professional
and Public Education of the American Heart Association Council on
High Blood pressure Research. Hypertension. 45:142–161.
2005.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Mancia G, De Backer G, Dominiczak A,
Cifkova R, Fagard R, Germano G, Grassi G, Heagerty AM, Kjeldsen SE,
Laurent S, et al: 2007 Guidelines for the management of arterial
hypertension: The Task Force for the Management of Arterial
Hypertension of the European Society of Hypertension (ESH) and of
the European Society of Cardiology (ESC). Eur Heart J.
28:1462–1536. 2007.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Ogihara T, Kikuchi K, Matsuoka H, Fujita
T, Higaki J, Horiuchi M, Imai Y, Imaizumi T, Ito S, Iwao H, et al:
The Japanese Society of Hypertension Guidelines for the Management
of Hypertension (JSH 2009). Hypertens Res. 32:3–107.
2009.PubMed/NCBI
|
|
46
|
Verdecchia P, Angeli F, Borgioni C,
Gattobigio R and Reboldi G: Ambulatory blood pressure and
cardiovascular outcome in relation to perceived sleep deprivation.
Hypertension. 49:777–783. 2007.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Henskens LH, van Boxtel MP, Kroon AA, van
Oostenbrugge RJ, Lodder J and de Leeuw PW: Subjective sleep
disturbance increases the nocturnal blood pressure level and
attenuates the correlation with target-organ damage. J Hypertens.
29:242–250. 2011.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Clement DL, De Buyzere ML, De Bacquer DA,
de Leeuw PW, Duprez DA, Fagard RH, Gheeraert PJ, Missault LH, Braun
JJ, Six RO, et al: Office versus Ambulatory Pressure Study
Investigators. Prognostic value of ambulatory blood-pressure
recordings in patients with treated hypertension. N Engl J Med.
348:2407–2415. 2003.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Dolan E, Stanton A, Thijs L, Hinedi K,
Atkins N, McClory S, Den Hond E, McCormack P, Staessen JA and
O'Brien E: Superiority of ambulatory over clinic blood pressure
measurement in predicting mortality: The Dublin outcome study.
Hypertension. 46:156–161. 2005.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Sega R, Facchetti R, Bombelli M, Cesana G,
Corrao G, Grassi G and Mancia G: Prognostic value of ambulatory and
home blood pressure compared with office blood pressure in the
general population: follow-up results from the Pressioni Arteriose
Monitorate e Loro Associazioni (PAMELA) study. Circulation.
111:1777–1783. 2005.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Ben-Dov IZ, Kark JD, Ben-Ishay D, Mekler
J, Ben-Arie L and Bursztyn M: Predictors of all-cause mortality in
clinical ambulatory monitoring: Unique aspects of blood pressure
during sleep. Hypertension. 49:1235–1241. 2007.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Boggia J, Li Y, Thijs L, Hansen TW, Kikuya
M, Bjorklund-Bodegard K, Richart T, Ohkubo T, Kuznetsova T,
Torp-Pedersen C, et al: Prognostic accuracy of day versus night
ambulatory blood pressure: A cohort study. Lancet. 370:1219–1229.
2007.PubMed/NCBI View Article : Google Scholar
|
|
53
|
O'Brien E, Sheridan J and O'Malley K:
Dippers and non-dippers. Lancet. 2(397)1988.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Verdecchia P, Schillaci G, Guerrieri M,
Gatteschi C, Benemio G, Boldrini F and Porcellati C: Circadian
blood pressure changes and left ventricular hypertrophy in
essential hypertension. Circulation. 81:528–536. 1990.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Shimada K, Kawamoto A, Matsubayashi K,
Nishinaga M, Kimura S and Ozawa T: Diurnal blood pressure
variations and silent cerebrovascular damage in elderly patients
with hypertension. J Hypertens. 10:875–878. 1992.PubMed/NCBI
|
|
56
|
Kario K, Matsuo T, Kobayashi H, Imiya M,
Matsuo M and Shimada K: Nocturnal fall of blood pressure and silent
cerebrovascular damage in elderly hypertensive patients. Advanced
silent cerebrovascular damage in extreme dippers. Hypertension.
27:130–135. 1996.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Umehara T, Toyoda C and Oka H:
Postprandial hypotension in de novo Parkinson's disease: A
comparison with orthostatic hypotension. Parkinsonism Relat Disord.
20:573–577. 2014.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Jansen RW and Lipsitz LA: Postprandial
hypotension: Epidemiology, pathophysiology, and clinical
management. Ann Intern Med. 122:286–295. 1995.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Thomaides T, Bleasdale-Barr K, Chaudhuri
KR, Pavitt D, Marsden CD and Mathias CJ: Cardiovascular and
hormonal responses to liquid food challenge in idiopathic
Parkinson's disease multiple system atrophy, and pure autonomic
failure. Neurology. 43:900–904. 1993.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Awosika A, Adabanya U, Millis RM, Omole AE
and Moon JH: Postprandial Hypotension: An underreported silent
killer in the aged. Cureus. 15(e35411)2023.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Low DA, da Nobrega AC and Mathias CJ:
Exercise-induced hypotension in autonomic disorders. Auton
Neurosci. 171:66–78. 2012.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Groom D: Cardiovascular observations on
Tarahumara Indian runners-the modern Spartans. Am Heart J.
81:304–314. 1971.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Smith GD and Mathias CJ: Postural
hypotension enhanced by exercise in patients with chronic autonomic
failure. QJM. 88:251–256. 1995.PubMed/NCBI
|
|
64
|
Kenney MJ and Seals DR: Postexercise
hypotension. Key features, mechanisms, and clinical significance.
Hypertension. 22:653–664. 1993.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Polito MD and Farinatti PT: The effects of
muscle mass and number of sets during resistance exercise on
postexercise hypotension. J Strength Cond Res. 23:2351–2357.
2009.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Chamsi-Pasha MA and Chamsi-Pasha H:
Avicenna's contribution to cardiology. Avicenna J Med. 4:9–12.
2014.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Nauman J, Janszky I, Vatten LJ and Wisloff
U: Temporal changes in resting heart rate and deaths from ischemic
heart disease. JAMA. 306:2579–2587. 2011.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Cole CR, Blackstone EH, Pashkow FJ, Snader
CE and Lauer MS: Heart-rate recovery immediately after exercise as
a predictor of mortality. N Engl J Med. 341:1351–1357.
1999.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Martin CE, Shaver JA, Leon DF, Thompson
ME, Reddy PS and Leonard JJ: Autonomic mechanisms in hemodynamic
responses to isometric exercise. J Clin Invest. 54:104–115.
1974.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Smith LL, Kukielka M and Billman GE: Heart
rate recovery after exercise: A predictor of ventricular
fibrillation susceptibility after myocardial infarction. Am J
Physiol Heart Circ Physiol. 288:H1763–H1769. 2005.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Hemingway H, Shipley M, Brunner E, Britton
A, Malik M and Marmot M: Does autonomic function link social
position to coronary risk? The Whitehall II study. Circulation.
111:3071–3077. 2005.PubMed/NCBI View Article : Google Scholar
|
|
72
|
La Rovere MT, Pinna GD, Maestri R, Mortara
A, Capomolla S, Febo O, Ferrari R, Franchini M, Gnemmi M, Opasich
C, et al: Short-term heart rate variability strongly predicts
sudden cardiac death in chronic heart failure patients.
Circulation. 107:565–570. 2003.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Maetzler W, Liepelt I and Berg D:
Progression of Parkinson's disease in the clinical phase: Potential
markers. Lancet Neurol. 8:1158–1171. 2009.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Miyagi T, Yamazato M, Nakamura T,
Tokashiki T, Namihira Y, Kokuba K, Ishihara S, Sakima H and Ohya Y:
Power spectral analysis of heart rate variability is useful as a
screening tool for detecting sympathetic and parasympathetic
nervous dysfunctions in Parkinson's disease. BMC Neurol.
22(339)2022.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Bidikar MP, Jagtap GJ and Chakor RT:
Influence of deep breathing on heart rate variability in
Parkinson's disease: Co-relation with severity of disease and
non-motor symptom scale score. J Clin Diagn Res. 8:BC01–BC03.
2014.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Kim JB, Phillips Z, Paik SH, Kang SY, Jeon
NJ, Kim BJ and Kim BM: Cerebral hemodynamic monitoring of
Parkinson's disease patients with orthostatic intolerance during
head-up tilt test. Neurophotonics. 7(025002)2020.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Schmidt C, Herting B, Prieur S, Junghanns
S, Schweitzer K, Globas C, Schöls L, Reichmann H, Berg D and
Ziemssen T: Valsalva manoeuvre in patients with different
Parkinsonian disorders. J Neural Transm (Vienna). 116:875–880.
2009.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Huang CC, Lai YR, Lien CY, Cheng BC, Kung
CT, Chiang YF and Lu CH: Effectiveness of different methods for
baroreflex sensitivity assessment in determining the severity of
cardiovascular autonomic neuropathy in patients With Parkinson's
disease. Front Neurosci. 16(833344)2022.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Puisieux F, Bulckaen H, Fauchais AL,
Drumez S, Salomez-Granier F and Dewailly P: Ambulatory blood
pressure monitoring and postprandial hypotension in elderly persons
with falls or syncopes. J Gerontol A Biol Sci Med Sci.
55:M535–M540. 2000.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Shen L, Yang X, Lu W, Chen W, Ye X and Wu
D: 24-hour ambulatory blood pressure alterations in patients with
Parkinson's disease. Brain Behav. 12(e2428)2022.PubMed/NCBI View Article : Google Scholar
|
|
81
|
DiFrancisco-Donoghue J, Elokda A, Lamberg
EM, Bono N and Werner WG: Norepinephrine and cardiovascular
responses to maximal exercise in Parkinson's disease on and off
medication. Mov Disord. 24:1773–1778. 2009.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Cohn JN: Plasma norepinephrine and
mortality. Clin Cardiol. 18 (3 Suppl I):I9–I12. 1995.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Bristow MR: Beta-adrenergic receptor
blockade in chronic heart failure. Circulation. 101:558–569.
2000.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Rowell LB, Johnson DG, Chase PB, Comess KA
and Seals DR: Hypoxemia raises muscle sympathetic activity but not
norepinephrine in resting humans. J Appl Physiol (1985).
66:1736–1743. 1989.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Akaogi Y, Asahina M, Yamanaka Y, Koyama Y
and Hattori T: Sudomotor, skin vasomotor, and cardiovascular
reflexes in 3 clinical forms of Lewy body disease. Neurology.
73:59–65. 2009.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Asahina M, Sakakibara R, Liu Z, Ito T,
Yamanaka Y, Nakazawa K, Shimizu E and Hattori T: The raphe
magnus/pallidus regulates sweat secretion and skin vasodilation of
the cat forepaw pad: A preliminary electrical stimulation study.
Neurosci Lett. 415:283–287. 2007.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Antonio-Rubio I, Madrid-Navarro CJ,
Salazar-Lopez E, Perez-Navarro MJ, Saez-Zea C, Gomez-Milan E,
Mínguez-Castellanos A and Escamilla-Sevilla F: Abnormal
thermography in Parkinson's disease. Park Relat Disord. 21:852–857.
2015.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Orimo S, Oka T, Miura H, Tsuchiya K, Mori
F, Wakabayashi K, Nagao T and Yokochi M: Sympathetic cardiac
denervation in Parkinson's disease and pure autonomic failure but
not in multiple system atrophy. J Neurol Neurosurg Psychiatry.
73:776–777. 2002.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Pitton Rissardo J and Fornari Caprara AL:
Cardiac 123I-Metaiodobenzylguanidine (MIBG) Scintigraphy in
Parkinson's disease: A comprehensive review. Brain Sci.
13(1471)2023.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Courbon F, Brefel-Courbon C, Thalamas C,
Alibelli MJ, Berry I, Montastruc JL, Rascol O and Senard JM:
Cardiac MIBG scintigraphy is a sensitive tool for detecting cardiac
sympathetic denervation in Parkinson's disease. Mov Disord.
18:890–897. 2003.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Kim JS, Park HE, Oh YS, Lee SH, Park JW,
Son BC and Lee KS: Orthostatic hypotension and cardiac sympathetic
denervation in Parkinson disease patients with REM sleep behavioral
disorder. J Neurol Sci. 362:59–63. 2016.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Yoshita M, Hayashi M and Hirai S:
Decreased myocardial accumulation of 123I-meta-iodobenzyl guanidine
in Parkinson's disease. Nucl Med Commun. 19:137–142.
1998.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Amino T, Orimo S, Itoh Y, Takahashi A,
Uchihara T and Mizusawa H: Profound cardiac sympathetic denervation
occurs in Parkinson disease. Brain Pathol. 15:29–34.
2005.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Orimo S, Ozawa E, Oka T, Nakade S,
Tsuchiya K, Yoshimoto M, Wakabayashi K and Takahashi H: Different
histopathology accounting for a decrease in myocardial MIBG uptake
in PD and MSA. Neurology. 57:1140–1141. 2001.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Bonuccelli U, Lucetti C, Del Dotto P,
Ceravolo R, Gambaccini G, Bernardini S, Rossi G and Piaggesi A:
Orthostatic hypotension in de novo Parkinson disease. Arch Neurol.
60:1400–1404. 2003.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Ejaz AA, Sekhon IS and Munjal S:
Characteristic findings on 24-h ambulatory blood pressure
monitoring in a series of patients with Parkinson's disease. Eur J
Intern Med. 17:417–420. 2006.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Kaufmann H and Goldstein DS: Autonomic
dysfunction in Parkinson disease. Handb Clin Neurol. 117:259–278.
2013.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Gibbons CH and Freeman R: Clinical
implications of delayed orthostatic hypotension: A 10-year
follow-up study. Neurology. 85:1362–1367. 2015.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Jamnadas-Khoda J, Koshy S, Mathias CJ,
Muthane UB, Ragothaman M and Dodaballapur SK: Are current
recommendations to diagnose orthostatic hypotension in Parkinson's
disease satisfactory? Mov Disord. 24:1747–1751. 2009.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Aarsland D, Andersen K, Larsen JP, Lolk A
and Kragh-Sorensen P: Prevalence and characteristics of dementia in
Parkinson disease: An 8-year prospective study. Arch Neurol.
60:387–392. 2003.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Merola A, Romagnolo A, Rosso M, Suri R,
Berndt Z, Maule S, Lopiano L and Espay AJ: Autonomic dysfunction in
Parkinson's disease: A prospective cohort study. Mov Disord.
33:391–397. 2018.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Longardner K, Bayram E and Litvan I:
Orthostatic hypotension is associated with cognitive decline in
Parkinson disease. Front Neurol. 11(897)2020.PubMed/NCBI View Article : Google Scholar
|
|
103
|
McDonald C, Newton JL and Burn DJ:
Orthostatic hypotension and cognitive impairment in Parkinson's
disease: Causation or association? Mov Disord. 31:937–946.
2016.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Palma JA and Kaufmann H: Treatment of
autonomic dysfunction in Parkinson disease and other
synucleinopathies. Mov Disord. 33:372–390. 2018.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Anang JB, Nomura T, Romenets SR, Nakashima
K, Gagnon JF and Postuma RB: Dementia Predictors in Parkinson
Disease: A validation study. J Parkinsons Dis. 7:159–162.
2017.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Cicero CE, Raciti L, Monastero R, Mostile
G, Donzuso G, Sciacca G, Luca A, Terravecchia C, Giuliano L, Baschi
R, et al: Cardiovascular autonomic function and MCI in Parkinson's
disease. Parkinsonism Relat Disord. 69:55–58. 2019.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Centi J, Freeman R, Gibbons CH, Neargarder
S, Canova AO and Cronin-Golomb A: Effects of Orthostatic
hypotension on cognition in Parkinson disease. Neurology. 88:17–24.
2017.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Sforza M, Assogna F, Rinaldi D, Sette G,
Tagliente S and Pontieri FE: Orthostatic hypotension acutely
impairs executive functions in Parkinson's disease. Neurol Sci.
39:1459–1462. 2018.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Peralta C, Stampfer-Kountchev M, Karner E,
Kollensperger M, Geser F, Wolf E, Seppi K, Benke T, Poewe W and
Wenning GK: Orthostatic hypotension and attention in Parkinson's
disease with and without dementia. J Neural Transm (Vienna).
114:585–588. 2007.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Fanciulli A, Gobel G, Ndayisaba JP,
Granata R, Duerr S, Strano S, Colosimo C, Poewe W, Pontieri FE and
Wenning GK: Supine hypertension in Parkinson's disease and multiple
system atrophy. Clin Auton Res. 26:97–105. 2016.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Jordan J and Biaggioni I: Diagnosis and
treatment of Supine hypertension in autonomic failure patients with
orthostatic hypotension. J Clin Hypertens Greenwich. 4:139–145.
2002.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Chobanian AV, Bakris GL, Black HR, Cushman
WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright
JT Jr, et al: The Seventh Report of the Joint National Committee on
prevention, detection, evaluation, and treatment of high blood
pressure: The JNC 7 report. JAMA. 289:2560–2572. 2003.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Kim JS, Oh YS, Lee KS, Kim YI, Yang DW and
Goldstein DS: Association of cognitive dysfunction with
neurocirculatory abnormalities in early Parkinson disease.
Neurology. 79:1323–1331. 2012.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Schmidt C, Berg D, Herting Prieur S,
Junghanns S, Schweitzer K, Globas C, Schöls L, Reichmann H and
Ziemssen T: Loss of nocturnal blood pressure fall in various
extrapyramidal syndromes. Mov Disord. 24:2136–2142. 2009.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Sommer S, Aral-Becher B and Jost W:
Nondipping in blood pressure. Park Dis. 2011(897586)2011.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Fanciulli A, Jordan J, Biaggioni I,
Calandra-Buonaura G, Cheshire WP, Cortelli P, Eschlboeck S, Grassi
G, Hilz MJ, Kaufmann H, et al: Consensus statement on the
definition of neurogenic SH in cardiovascular autonomic failure by
the American Autonomic Society (AAS) and the European Federation of
Autonomic Societies (EFAS) : Endorsed by the European Academy of
Neurology (EAN) and the European Society of Hypertension (ESH).
Clin Auton Res. 28:355–362. 2018.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Umehara T, Matsuno H, Toyoda C and Oka H:
Clinical characteristics of Supine hypertension in de novo
Parkinson disease. Clin Auton Res. 26:15–21. 2016.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Palma JA, Redel-Traub G, Porciuncula A,
Samaniego-Toro D, Millar Vernetti P, Lui YW, Norcliffe-Kaufmann L
and Kaufmann H: The impact of Supine hypertension on target organ
damage and survival in patients with synucleinopathies and
neurogenic orthostatic hypotension. Park Relat Disord. 75:97–104.
2020.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Tanaka R, Shimo Y, Yamashiro K, Ogawa T,
Nishioka K, Oyama G, Umemura A and Hattori N: Association between
abnormal nocturnal blood pressure profile and dementia in
Parkinson's disease. Parkinsonism Relat Disord. 46:24–29.
2018.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Oh YS, Kim JS, Yang DW, Koo JS, Kim YI,
Jung HO and Lee KS: Nighttime blood pressure and white matter
hyperintensities in patients with Parkinson disease. Chronobiol
Int. 30:811–817. 2013.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Sunwoo MK, Jeon S, Ham JH, Hong JY, Lee
JE, Lee JM, Sohn YH and Lee PH: The burden of white matter
hyperintensities is a predictor of progressive mild cognitive
impairment in patients with Parkinson's disease. Eur J Neurol.
21:922–e50. 2014.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Oka H, Umehara T, Nakahara A and Matsuno
H: Comparisons of cardiovascular dysautonomia and cognitive
impairment between de novo Parkinson's disease and de novo dementia
with Lewy bodies. BMC Neurol. 20(350)2020.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Fengler S, Liepelt-Scarfone I, Brockmann
K, Schaffer E, Berg D and Kalbe E: Cognitive changes in prodromal
Parkinson's disease: A review. Mov Disord. 32:1655–1666.
2017.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Perez F, Helmer C, Foubert-Samier A,
Auriacombe S, Dartigues JF and Tison F: Risk of dementia in an
elderly population of Parkinson's disease patients: A 15-year
population-based study. Alzheimers Dement. 8:463–469.
2012.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Aarsland D, Andersen K, Larsen JP, Lolk A,
Nielsen H and Kragh-Sorensen P: Risk of dementia in Parkinson's
disease: A community-based, prospective study. Neurology.
56:730–736. 2001.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Aarsland D, Zaccai J and Brayne C: A
systematic review of prevalence studies of dementia in Parkinson's
disease. Mov Disord. 20:1255–1263. 2005.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Aarsland D, Larsen JP, Tandberg E and
Laake K: Predictors of nursing home placement in Parkinson's
disease: A population-based, prospective study. J Am Geriatr Soc.
48:938–942. 2000.PubMed/NCBI View Article : Google Scholar
|
|
128
|
McKeith IG, Ferman TJ, Thomas AJ, Blanc F,
Boeve BF, Fujishiro H, Kantarci K, Muscio C, O'Brien JT, Postuma
RB, et al: Research criteria for the diagnosis of prodromal
dementia with Lewy bodies. Neurology. 94:743–755. 2020.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Aarsland D, Bronnick K, Williams-Gray C,
Weintraub D, Marder K, Kulisevsky J, Burn D and Barone P: Mild
cognitive impairment in Parkinson disease: A multicenter pooled
analysis. Neurology. 75:1062–1069. 2010.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Lawson RA, Yarnall AJ, Duncan GW, Breen
DP, Khoo TK, Williams-Gray CH, Barker RA and Burn DJ: ICICLE-PD
study group. Stability of mild cognitive impairment in newly
diagnosed Parkinson's disease. J Neurol Neurosurg Psychiatry.
88:648–652. 2017.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Domellof ME, Ekman U, Forsgren L and Elgh
E: Cognitive function in the early phase of Parkinson's disease, a
five-year follow-up. Acta Neurol Scand. 132:79–88. 2015.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Weintraub D, Simuni T, Caspell-Garcia C,
Coffey C, Lasch S, Siderowf A, Aarsland D, Barone P and Burn D:
Cognitive performance and neuropsychiatric symptoms in early,
untreated Parkinson's disease. Mov Disord. 30:919–927.
2015.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Pedersen KF, Larsen JP, Tysnes OB and
Alves G: Natural course of mild cognitive impairment in Parkinson
disease: A 5-year population-based study. Neurology. 88:767–774.
2017.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Jones JD, Kuhn TP and Szymkowicz SM:
Reverters from PD-MCI to cognitively intact are at risk for future
cognitive impairment: Analysis of the PPMI cohort. Parkinsonism
Relat Disord. 47:3–7. 2018.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Erro R, Santangelo G, Barone P, Picillo M,
Amboni M, Longo K, Giordano F, Moccia M, Allocca R, Pellecchia MT
and Vitale C: Do subjective memory complaints herald the onset of
mild cognitive impairment in Parkinson disease? J Geriatr
Psychiatry Neurol. 27:276–281. 2014.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Purri R, Brennan L, Rick J, Xie SX, Deck
BL, Chahine LM, Dahodwala N, Chen-Plotkin A, Duda JE, Morley JF, et
al: Subjective Cognitive Complaint in Parkinson's disease patients
with normal cognition: Canary in the coal mine? Mov Disord.
35:1618–1625. 2020.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Chua CY, Koh MRE, Chia NS, Ng SY, Saffari
SE, Wen MC, Chen RY, Choi X, Heng DL, Neo SX, et al: Subjective
cognitive Complaints in early Parkinson's disease patients with
normal cognition are associated with affective symptoms.
Parkinsonism Relat Disord. 82:24–28. 2021.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Szewczyk-Krolikowski K, Tomlinson P, Nithi
K, Wade-Martins R, Talbot K, Ben-Shlomo Y and Hu MT: The influence
of age and gender on motor and non-motor features of early
Parkinson's disease: Initial findings from the Oxford Parkinson
Disease Center (OPDC) discovery cohort. Parkinsonism Relat Disord.
20:99–105. 2014.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Augustine EF, Perez A, Dhall R, Umeh CC,
Videnovic A, Cambi F, Wills AM, Elm JJ, Zweig RM, Shulman LM, et
al: Sex differences in clinical features of early, treated
Parkinson's disease. PLoS One. 10(e0133002)2015.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Gao L, Nie K, Tang H, Wang L, Zhao J, Gan
R, Huang J, Feng S, Zhu R, Duan Z, et al: Sex differences in
cognition among Chinese people with Parkinson's disease. J Clin
Neurosci. 22:488–492. 2015.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Liu R, Umbach DM, Peddada SD, Xu Z,
Troster AI, Huang X and Chen H: Potential sex differences in
nonmotor symptoms in early drug-naive Parkinson disease. Neurology.
84:2107–2115. 2015.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Sundermann EE, Maki PM, Rubin LH, Lipton
RB, Landau S and Biegon A: Alzheimer's Disease Neuroimaging
Initiative. Female advantage in verbal memory: Evidence of
sex-specific cognitive reserve. Neurology. 87:1916–1924.
2016.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Gonzalez-Latapi P, Bayram E, Litvan I and
Marras C: Cognitive Impairment in Parkinson's disease:
Epidemiology, clinical profile, protective and risk factors. Behav
Sci (Basel). 11(74)2021.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Compta Y, Valente T, Saura J, Segura B,
Iranzo A, Serradell M, Junqué C, Tolosa E, Valldeoriola F, Muñoz E,
et al: Correlates of cerebrospinal fluid levels of oligomeric- and
total-α-synuclein in premotor, motor and dementia stages of
Parkinson's disease. J Neurol. 262:294–306. 2015.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Stewart T, Liu C, Ginghina C, Cain KC,
Auinger P, Cholerton B, Shi M and Zhang J: Parkinson Study Group
DATATOP Investigators. Cerebrospinal fluid α-synuclein predicts
cognitive decline in Parkinson disease progression in the DATATOP
cohort. Am J Pathol. 184:966–975. 2014.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Hansson O, Hall S, Ohrfelt A, Zetterberg
H, Blennow K, Minthon L, Nägga K, Londos E, Varghese S, Majbour NK,
et al: Levels of cerebrospinal fluid α-synuclein oligomers are
increased in Parkinson's disease with dementia and dementia with
Lewy bodies compared to Alzheimer's disease. Alzheimers Res Ther.
6(25)2014.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Coughlin DG, Hurtig HI and Irwin DJ:
Pathological influences on clinical heterogeneity in lewy body
diseases. Mov Disord. 35:5–19. 2020.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Robinson JL, Lee EB, Xie SX, Rennert L,
Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, et
al: Neurodegenerative disease concomitant proteinopathies are
prevalent, age-related and APOE4-associated. Brain. 141:2181–2193.
2018.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Smith C, Malek N, Grosset K, Cullen B,
Gentleman S and Grosset DG: Neuropathology of dementia in patients
with Parkinson's disease: A systematic review of autopsy studies. J
Neurol Neurosurg Psychiatry. 90:1234–1243. 2019.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Mollenhauer B, Trenkwalder C, von Ahsen N,
Bibl M, Steinacker P, Brechlin P, Schindehuette J, Poser S,
Wiltfang J and Otto M: Beta-amlyoid 1-42 and tau-protein in
cerebrospinal fluid of patients with Parkinson's disease dementia.
Dement Geriatr Cogn Disord. 22:200–208. 2006.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Compta Y, Pereira JB, Rios J,
Ibarretxe-Bilbao N, Junque C, Bargallo N, Cámara A, Buongiorno M,
Fernández M, Pont-Sunyer C and Martí MJ: Combined dementia-risk
biomarkers in Parkinson's disease: A prospective longitudinal
study. Parkinsonism Relat Disord. 19:717–724. 2013.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Howlett DR, Whitfield D, Johnson M, Attems
J, O'Brien JT, Aarsland D, Lai MK, Lee JH, Chen C, Ballard C, et
al: Regional multiple pathology scores are associated with
cognitive decline in lewy body dementias. Brain Pathol. 25:401–408.
2015.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Jellinger KA and Attems J: Cerebral
amyloid angiopathy in Lewy body disease. J Neural Transm (Vienna).
115:473–482. 2008.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Hansen D, Ling H, Lashley T, Foley JA,
Strand C, Eid TM, Holton JL and Warner TT: Novel
clinicopathological characteristics differentiate dementia with
Lewy bodies from Parkinson's disease dementia. Neuropathol Appl
Neurobiol. 47:143–156. 2021.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Chahine LM, Xie SX, Simuni T, Tran B,
Postuma R, Amara A, Oertel WH, Iranzo A, Scordia C, Fullard M, et
al: Longitudinal changes in cognition in early Parkinson's disease
patients with REM sleep behavior disorder. Parkinsonism Relat
Disord. 27:102–106. 2016.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Forbes E, Tropea TF, Mantri S, Xie SX and
Morley JF: Modifiable comorbidities associated with cognitive
decline in Parkinson's disease. Mov Disord Clin Pract. 8:254–263.
2021.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Guo Y, Xu W, Liu FT, Li JQ, Cao XP, Tan L,
Wang J and Yu JT: Modifiable risk factors for cognitive impairment
in Parkinson's disease: A systematic review and meta-analysis of
prospective cohort studies. Mov Disord. 34:876–883. 2019.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Kivipelto M, Ngandu T, Laatikainen T,
Winblad B, Soininen H and Tuomilehto J: Risk score for the
prediction of dementia risk in 20 years among middle aged people: A
longitudinal, population-based study. Lancet Neurol. 5:735–741.
2006.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Chahine LM, Dos Santos C, Fullard M,
Scordia C, Weintraub D, Erus G, Rosenthal L, Davatzikos C and
McMillan CT: Modifiable vascular risk factors, white matter disease
and cognition in early Parkinson's disease. Eur J Neurol.
26:246–e18. 2019.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Dong C, Nabizadeh N, Caunca M, Cheung YK,
Rundek T, Elkind MS, DeCarli C, Sacco RL, Stern Y and Wright CB:
Cognitive correlates of white matter lesion load and brain atrophy:
The Northern Manhattan Study. Neurology. 85:441–449.
2015.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Erten-Lyons D, Woltjer R, Kaye J, Mattek
N, Dodge HH, Green S, Tran H, Howieson DB, Wild K and Silbert LC:
Neuropathologic basis of white matter hyperintensity accumulation
with advanced age. Neurology. 81:977–983. 2013.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Mollenhauer B, Zimmermann J, Sixel-Doring
F, Focke NK, Wicke T, Ebentheuer J, Schaumburg M, Lang E, Friede T
and Trenkwalder C: DeNoPa Study Group. Baseline predictors for
progression 4 years after Parkinson's disease diagnosis in the De
Novo Parkinson Cohort (DeNoPa). Mov Disord. 34:67–77.
2019.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Kim HJ, Oh ES, Lee JH, Moon JS, Oh JE,
Shin JW, Lee KJ, Baek IC, Jeong SH, Song HJ, et al: Relationship
between changes of body mass index (BMI) and cognitive decline in
Parkinson's disease (PD). Arch Gerontol Geriatr. 55:70–72.
2012.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Yoo HS, Chung SJ, Lee PH, Sohn YH and Kang
SY: The influence of body mass index at diagnosis on cognitive
decline in Parkinson's disease. J Clin Neurol. 15:517–526.
2019.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Anang JB, Gagnon JF, Bertrand JA, Romenets
SR, Latreille V, Panisset M, Montplaisir J and Postuma RB:
Predictors of dementia in Parkinson disease: A prospective cohort
study. Neurology. 83:1253–1260. 2014.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Goldstein DS, Holmes CS, Dendi R, Bruce SR
and Li ST: Orthostatic hypotension from sympathetic denervation in
Parkinson's disease. Neurology. 58:1247–1255. 2002.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Papapetropoulos S, Gonzalez J and Mash DC:
The effect of ischemic cerebrovascular disease on the clinical
characteristics of Parkinson's disease. A post-mortem study. Eur J
Neurol. 13:96–97. 2006.PubMed/NCBI View Article : Google Scholar
|
|
168
|
Moran LB and Graeber MB: Towards a pathway
definition of Parkinson's disease: A complex disorder with links to
cancer, diabetes and inflammation. Neurogenetics. 9:1–13.
2008.PubMed/NCBI View Article : Google Scholar
|
|
169
|
Menon R and Farina C: Shared molecular and
functional frameworks among five complex human disorders: A
comparative study on interactomes linked to susceptibility genes.
PLoS One. 6(e18660)2011.PubMed/NCBI View Article : Google Scholar
|
|
170
|
Cheong JLY, de Pablo-Fernandez E, Foltynie
T and Noyce AJ: The association between type 2 diabetes mellitus
and Parkinson's disease. J Parkinsons Dis. 10:775–789.
2020.PubMed/NCBI View Article : Google Scholar
|
|
171
|
Santiago JA and Potashkin JA: Shared
dysregulated pathways lead to Parkinson's disease and diabetes.
Trends Mol Med. 19:176–186. 2013.PubMed/NCBI View Article : Google Scholar
|
|
172
|
Bosco D, Plastino M, Cristiano D, Colica
C, Ermio C, De Bartolo M, Mungari P, Fonte G, Consoli D, Consoli A
and Fava A: Dementia is associated with insulin resistance in
patients with Parkinson's disease. J Neurol Sci. 315:39–43.
2012.PubMed/NCBI View Article : Google Scholar
|
|
173
|
Shen C, Guo Y, Luo W, Lin C and Ding M:
Serum urate and the risk of Parkinson's disease: Results from a
meta-analysis. Can J Neurol Sci. 40:73–79. 2013.PubMed/NCBI View Article : Google Scholar
|
|
174
|
Weisskopf MG, O'Reilly E, Chen H,
Schwarzschild MA and Ascherio A: Plasma urate and risk of
Parkinson's disease. Am J Epidemiol. 166:561–567. 2007.PubMed/NCBI View Article : Google Scholar
|
|
175
|
Jain S, Ton TG, Boudreau RM, Yang M,
Thacker EL, Studenski S, Longstreth WT Jr, Strotmeyer ES and Newman
AB: The risk of Parkinson disease associated with urate in a
community-based cohort of older adults. Neuroepidemiology.
36:223–229. 2011.PubMed/NCBI View Article : Google Scholar
|
|
176
|
Rissardo JP and Caprara ALF: Uric acid and
Parkinson's disease. Menoufia Med J. 35:2093–2094. 2023.
|
|
177
|
Maetzler W, Stapf AK, Schulte C, Hauser
AK, Lerche S, Wurster I, Schleicher E, Melms A and Berg D: Serum
and cerebrospinal fluid uric acid levels in lewy body disorders:
associations with disease occurrence and amyloid-β pathway. J
Alzheimers Dis. 27:119–126. 2011.PubMed/NCBI View Article : Google Scholar
|
|
178
|
Annanmaki T, Pessala-Driver A, Hokkanen L
and Murros K: Uric acid associates with cognition in Parkinson's
disease. Parkinsonism Relat Disord. 14:576–578. 2008.PubMed/NCBI View Article : Google Scholar
|
|
179
|
Moccia M, Picillo M, Erro R, Vitale C,
Longo K, Amboni M, Santangelo G, Spina E, De Rosa A, De Michele G,
et al: Is serum uric acid related to non-motor symptoms in de-novo
Parkinson's disease patients? Parkinsonism Relat Disord.
20:772–775. 2014.PubMed/NCBI View Article : Google Scholar
|
|
180
|
Moccia M, Picillo M, Erro R, Vitale C,
Longo K, Amboni M, Santangelo G, Palladino R, Capo G, Orefice G, et
al: Presence and progression of non-motor symptoms in relation to
uric acid in de novo Parkinson's disease. Eur J Neurol. 22:93–98.
2015.PubMed/NCBI View Article : Google Scholar
|
|
181
|
Block ML and Hong JS: Microglia and
inflammation-mediated neurodegeneration: Multiple triggers with a
common mechanism. Prog Neurobiol. 76:77–98. 2005.PubMed/NCBI View Article : Google Scholar
|
|
182
|
Fan Z, Aman Y, Ahmed I, Chetelat G,
Landeau B, Ray Chaudhuri K, Brooks DJ and Edison P: Influence of
microglial activation on neuronal function in Alzheimer's and
Parkinson's disease dementia. Alzheimers Dement. 11:608–621.e7.
2015.PubMed/NCBI View Article : Google Scholar
|
|
183
|
Aviles-Olmos I, Limousin P, Lees A and
Foltynie T: Parkinson's disease, insulin resistance and novel
agents of neuroprotection. Brain. 136(Pt 2):374–384.
2013.PubMed/NCBI View Article : Google Scholar
|
|
184
|
Garabadu D, Agrawal N, Sharma A and Sharma
S: Mitochondrial metabolism: A common link between
neuroinflammation and neurodegeneration. Behav Pharmacol.
30:642–652. 2019.PubMed/NCBI View Article : Google Scholar
|
|
185
|
Jeon YM, Kwon Y, Jo M, Lee S, Kim S and
Kim HJ: The Role Glial Mitochondria in α-Synuclein Toxicity. Front
Cell Dev Biol. 8(548283)2020.PubMed/NCBI View Article : Google Scholar
|
|
186
|
Gatt AP, Duncan OF, Attems J, Francis PT,
Ballard CG and Bateman JM: Dementia in Parkinson's disease is
associated with enhanced mitochondrial complex I deficiency. Mov
Disord. 31:352–359. 2016.PubMed/NCBI View Article : Google Scholar
|
|
187
|
Toklu HZ and Tumer N: Oxidative Stress,
Brain Edema, Blood-Brain Barrier Permeability, and Autonomic
Dysfunction from Traumatic Brain Injury. In: Brain Neurotrauma:
Molecular, Neuropsychological, and Rehabilitation Aspects. Kobeissy
FH (Ed.). Chapter 5. CRC Press/Taylor & Francis, Boca Raton Fl,
2015.
|
|
188
|
Krauss JK: Movement disorders secondary to
craniocerebral trauma. Handb Clin Neurol. 128:475–496.
2015.PubMed/NCBI View Article : Google Scholar
|
|
189
|
Schiehser DM, Filoteo JV, Litvan I,
Pirogovsky-Turk E, Lessig SL and Song DS: Cognitive functioning in
individuals with Parkinson's disease and traumatic brain injury: A
longitudinal study. Parkinsonism Relat Disord. 30:58–61.
2016.PubMed/NCBI View Article : Google Scholar
|
|
190
|
Nombela C, Rowe JB, Winder-Rhodes SE,
Hampshire A, Owen AM, Breen DP, Duncan GW, Khoo TK, Yarnall AJ,
Firbank MJ, et al: Genetic impact on cognition and brain function
in newly diagnosed Parkinson's disease: ICICLE-PD Study. Brain.
137(Pt 10):2743–2758. 2014.PubMed/NCBI View Article : Google Scholar
|
|
191
|
Paul KC, Rausch R, Creek MM, Sinsheimer
JS, Bronstein JM, Bordelon Y and Ritz B: APOE, MAPT, and COMT and
Parkinson's disease susceptibility and cognitive symptom
progression. J Park Dis. 6:349–359. 2016.PubMed/NCBI View Article : Google Scholar
|
|
192
|
Williams-Gray CH, Goris A, Saiki M,
Foltynie T, Compston DA, Sawcer SJ and Barker RA: Apolipoprotein E
genotype as a risk factor for susceptibility to and dementia in
Parkinson's disease. J Neurol. 256:493–498. 2009.PubMed/NCBI View Article : Google Scholar
|
|
193
|
Williams-Gray CH, Mason SL, Evans JR,
Foltynie T, Brayne C, Robbins TW and Barker RA: The CamPaIGN study
of Parkinson's disease: 10-year outlook in an incident
population-based cohort. J Neurol Neurosurg Psychiatry.
84:1258–1264. 2013.PubMed/NCBI View Article : Google Scholar
|
|
194
|
Harward SC, Hedrick NG, Hall CE,
Parra-Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R
and McNamara JO: Autocrine BDNF-TrkB signalling within a single
dendritic spine. Nature. 538:99–103. 2016.PubMed/NCBI View Article : Google Scholar
|
|
195
|
Chen ZY, Patel PD, Sant G, Meng CX, Teng
KK, Hempstead BL and Lee FS: Variant brain-derived neurotrophic
factor (BDNF) (Met66) alters the intracellular trafficking and
activity-dependent secretion of wild-type BDNF in neurosecretory
cells and cortical neurons. J Neurosci. 24:4401–4411.
2004.PubMed/NCBI View Article : Google Scholar
|
|
196
|
Guerini FR, Beghi E, Riboldazzi G,
Zangaglia R, Pianezzola C, Bono G, Casali C, Di Lorenzo C, Agliardi
C, Nappi G, et al: BDNF Val66Met polymorphism is associated with
cognitive impairment in Italian patients with Parkinson's disease.
Eur J Neurol. 16:1240–1245. 2009.PubMed/NCBI View Article : Google Scholar
|
|
197
|
Karakasis C, Kalinderi K, Katsarou Z,
Fidani L and Bostantjopoulou S: Association of brain-derived
neurotrophic factor (BDNF) Val66Met polymorphism with Parkinson's
disease in a Greek population. J Clin Neurosci. 18:1744–1745.
2011.PubMed/NCBI View Article : Google Scholar
|
|
198
|
The CA, Lee TS, Kuchibhatla M, Ashley-Koch
A, Macfall J, Krishnan R and Beyer J: Bipolar disorder,
brain-derived neurotrophic factor (BDNF) Val66Met polymorphism and
brain morphology. PLoS One. 7(e38469)2012.PubMed/NCBI View Article : Google Scholar
|
|
199
|
Svetel M, Pekmezovic T, Markovic V,
Novakovic I, Dobricic V, Djuric G, Stefanova E and Kostić V: No
association between brain-derived neurotrophic factor G196A
polymorphism and clinical features of Parkinson's disease. Eur
Neurol. 70:257–262. 2013.PubMed/NCBI View Article : Google Scholar
|
|
200
|
Poewe W, Seppi K, Tanner CM, Halliday GM,
Brundin P, Volkmann J, Schrag AE and Lang AE: Parkinson disease.
Nat Rev Primer. 3(17013)2017.PubMed/NCBI View Article : Google Scholar
|
|
201
|
Bridi JC and Hirth F: Mechanisms of
α-Synuclein Induced Synaptopathy in Parkinson's disease. Front
Neurosci. 12(80)2018.PubMed/NCBI View Article : Google Scholar
|
|
202
|
Aarsland D, Batzu L, Halliday GM, Geurtsen
GJ, Ballard C, Ray Chaudhuri K and Weintraub D: Parkinson
disease-associated cognitive impairment. Nat Rev Primer.
7(47)2021.PubMed/NCBI View Article : Google Scholar
|
|
203
|
Sasikumar S and Strafella AP: Imaging Mild
Cognitive Impairment and Dementia in Parkinson's disease. Front
Neurol. 11(47)2020.PubMed/NCBI View Article : Google Scholar
|
|
204
|
Ranganath A and Jacob SN: Doping the Mind:
Dopaminergic modulation of prefrontal cortical cognition.
Neuroscientist. 22:593–603. 2016.PubMed/NCBI View Article : Google Scholar
|
|
205
|
Westbrook A, van den Bosch R, Maatta JI,
Hofmans L, Papadopetraki D, Cools R and Frank MJ: Dopamine promotes
cognitive effort by biasing the benefits versus costs of cognitive
work. Science. 367:1362–1366. 2020.PubMed/NCBI View Article : Google Scholar
|
|
206
|
Keren NI, Taheri S, Vazey EM, Morgan PS,
Granholm AC, Aston-Jones GS and Eckert MA: Histologic validation of
locus coeruleus MRI contrast in post-mortem tissue. Neuroimage.
113:235–245. 2015.PubMed/NCBI View Article : Google Scholar
|
|
207
|
Borodovitsyna O, Flamini M and Chandler D:
Noradrenergic modulation of cognition in health and disease. Neural
Plast. 2017(6031478)2017.PubMed/NCBI View Article : Google Scholar
|
|
208
|
Ehrminger M, Latimier A, Pyatigorskaya N,
Garcia-Lorenzo D, Leu-Semenescu S, Vidailhet M, Lehericy S and
Arnulf I: The coeruleus/subcoeruleus complex in idiopathic rapid
eye movement sleep behaviour disorder. Brain. 139(Pt 4):1180–1188.
2016.PubMed/NCBI View Article : Google Scholar
|
|
209
|
Sommerauer M, Fedorova TD, Hansen AK,
Knudsen K, Otto M, Jeppesen J, Frederiksen Y and Blicher JU:
Evaluation of the noradrenergic system in Parkinson's disease: An
11C-MeNER PET and neuromelanin MRI study. Brain. 141:496–504.
2018.PubMed/NCBI View Article : Google Scholar
|
|
210
|
Sinn DI and Gibbons CH: Pathophysiology
and Treatment of Orthostatic Hypotension in Parkinsonian Disorders.
Curr Treat Options Neurol. 18(28)2016.PubMed/NCBI View Article : Google Scholar
|
|
211
|
Zhan Y, Raza MU, Yuan L and Zhu MY:
Critical Role of Oxidatively Damaged DNA in Selective Noradrenergic
Vulnerability. Neuroscience. 422:184–201. 2019.PubMed/NCBI View Article : Google Scholar
|
|
212
|
Buddhala C, Loftin SK, Kuley BM, Cairns
NJ, Campbell MC, Perlmutter JS and Kotzbauer PT: Dopaminergic,
serotonergic, and noradrenergic deficits in Parkinson disease. Ann
Clin Transl Neurol. 2:949–959. 2015.PubMed/NCBI View Article : Google Scholar
|
|
213
|
Betts MJ, Kirilina E, Otaduy MCG, Ivanov
D, Acosta-Cabronero J, Callaghan MF, Lambert C, Cardenas-Blanco A,
Pine K, Passamonti L, et al: Locus coeruleus imaging as a biomarker
for noradrenergic dysfunction in neurodegenerative diseases. Brain.
142:2558–2571. 2019.PubMed/NCBI View Article : Google Scholar
|
|
214
|
Schulz J, Pagano G, Fernandez Bonfante JA,
Wilson H and Politis M: Nucleus basalis of Meynert degeneration
precedes and predicts cognitive impairment in Parkinson's disease.
Brain. 141:1501–1516. 2018.PubMed/NCBI View Article : Google Scholar
|
|
215
|
Bohnen NI, Albin RL, Muller ML, Petrou M,
Kotagal V, Koeppe RA, Scott PJ and Frey KA: Frequency of
cholinergic and caudate nucleus dopaminergic deficits across the
predemented cognitive spectrum of Parkinson disease and evidence of
interaction effects. JAMA Neurol. 72:194–200. 2015.PubMed/NCBI View Article : Google Scholar
|
|
216
|
Pereira JB, Hall S, Jalakas M, Grothe MJ,
Strandberg O, Stomrud E, Westman E, van Westen D and Hansson O:
Longitudinal degeneration of the basal forebrain predicts
subsequent dementia in Parkinson's disease. Neurobiol Dis.
139(104831)2020.PubMed/NCBI View Article : Google Scholar
|
|
217
|
Ballinger EC, Ananth M, Talmage DA and
Role LW: Basal forebrain cholinergic circuits and signaling in
cognition and cognitive decline. Neuron. 91:1199–1218.
2016.PubMed/NCBI View Article : Google Scholar
|
|
218
|
Pillet LE, Taccola C, Cotoni J, Thiriez H,
Andre K and Verpillot R: Correction: Correlation between cognition
and plasma noradrenaline level in Alzheimer's disease: A potential
new blood marker of disease evolution. Transl Psychiatry.
10(409)2020.PubMed/NCBI View Article : Google Scholar
|
|
219
|
Alexandris AS, Walker L, Liu AKL, McAleese
KE, Johnson M, Pearce RKB, Gentleman SM and Attems J: Cholinergic
deficits and galaninergic hyperinnervation of the nucleus basalis
of Meynert in Alzheimer's disease and Lewy body disorders.
Neuropathol Appl Neurobiol. 46:264–278. 2020.PubMed/NCBI View Article : Google Scholar
|
|
220
|
Mattila PM, Roytta M, Lonnberg P,
Marjamaki P, Helenius H and Rinne JO: Choline acetytransferase
activity and striatal dopamine receptors in Parkinson's disease in
relation to cognitive impairment. Acta Neuropathol. 102:160–166.
2001.PubMed/NCBI View Article : Google Scholar
|
|
221
|
Gaykema RP and Zaborszky L: Direct
catecholaminergic-cholinergic interactions in the basal forebrain.
II. Substantia nigra-ventral tegmental area projections to
cholinergic neurons. J Comp Neurol. 374:555–577. 1996.PubMed/NCBI View Article : Google Scholar
|
|
222
|
Gargouri F, Gallea C, Mongin M,
Pyatigorskaya N, Valabregue R, Ewenczyk C, Sarazin M, Yahia-Cherif
L, Vidailhet M and Lehéricy S: Multimodal magnetic resonance
imaging investigation of basal forebrain damage and cognitive
deficits in Parkinson's disease. Mov Disord. 34:516–525.
2019.PubMed/NCBI View Article : Google Scholar
|
|
223
|
Hall H, Reyes S, Landeck N, Bye C, Leanza
G, Double K, Thompson L, Halliday G and Kirik D: Hippocampal Lewy
pathology and cholinergic dysfunction are associated with dementia
in Parkinson's disease. Brain. 137(Pt 9):2493–2508. 2014.PubMed/NCBI View Article : Google Scholar
|
|
224
|
Liu AKL, Chau TW, Lim EJ, Ahmed I, Chang
RC, Kalaitzakis ME, Graeber MB, Gentleman SM and Pearce RKB:
Hippocampal CA2 Lewy pathology is associated with cholinergic
degeneration in Parkinson's disease with cognitive decline. Acta
Neuropathol Commun. 7(61)2019.PubMed/NCBI View Article : Google Scholar
|
|
225
|
Nicastro N, Garibotto V and Burkhard PR:
Extrastriatal 123I-FP-CIT SPECT impairment in
Parkinson's disease - the PPMI cohort. BMC Neurol.
20(192)2020.PubMed/NCBI View Article : Google Scholar
|
|
226
|
Qamhawi Z, Towey D, Shah B, Pagano G,
Seibyl J, Marek K, Borghammer P, Brooks DJ and Pavese N: Clinical
correlates of raphe serotonergic dysfunction in early Parkinson's
disease. Brain. 138 (Pt 10):2964–2973. 2015.PubMed/NCBI View Article : Google Scholar
|
|
227
|
Maillet A, Krack P, Lhommee E, Metereau E,
Klinger H, Favre E, Le Bars D, Schmitt E, Bichon A, Pelissier P, et
al: The prominent role of serotonergic degeneration in apathy,
anxiety and depression in de novo Parkinson's disease. Brain. 139
(Pt 9):2486–2502. 2016.PubMed/NCBI View Article : Google Scholar
|
|
228
|
Smith CR, Cullen B, Sheridan MP, Cavanagh
J, Grosset KA and Grosset DG: Cognitive impairment in Parkinson's
disease is multifactorial: A neuropsychological study. Acta Neurol
Scand. 141:500–508. 2020.PubMed/NCBI View Article : Google Scholar
|
|
229
|
Amin J, Holmes C, Dorey RB, Tommasino E,
Casal YR, Williams DM, Dupuy C, Nicoll JAR and Boche D:
Neuroinflammation in dementia with Lewy bodies: A human post-mortem
study. Transl Psychiatry. 10(267)2020.PubMed/NCBI View Article : Google Scholar
|
|
230
|
Devanand DP, Pradhaban G, Liu X, Khandji
A, De Santi S, Segal S, Rusinek H, Pelton GH, Honig LS, Mayeux R,
et al: Hippocampal and entorhinal atrophy in mild cognitive
impairment: Prediction of Alzheimer disease. Neurology. 68:828–836.
2007.PubMed/NCBI View Article : Google Scholar
|
|
231
|
Harding AJ and Halliday GM: Cortical Lewy
body pathology in the diagnosis of dementia. Acta Neuropathol.
102:355–363. 2001.PubMed/NCBI View Article : Google Scholar
|
|
232
|
Schaser AJ, Osterberg VR, Dent SE,
Stackhouse TL, Wakeham CM, Boutros SW, Weston LJ, Owen N, Weissman
TA, Luna E, et al: Alpha-synuclein is a DNA binding protein that
modulates DNA repair with implications for Lewy body disorders. Sci
Rep. 9(10919)2019.PubMed/NCBI View Article : Google Scholar
|
|
233
|
Petrou M, Dwamena BA, Foerster BR,
MacEachern MP, Bohnen NI, Muller ML, Albin RL and Frey KA: Amyloid
deposition in Parkinson's disease and cognitive impairment: A
systematic review. Mov Disord. 30:928–935. 2015.PubMed/NCBI View Article : Google Scholar
|
|
234
|
Reimand J, Boon BDC, Collij LE, Teunissen
CE, Rozemuller AJM, van Berckel BNM, Scheltens P, Ossenkoppele R
and Bouwman F: Amyloid-β PET and CSF in an autopsy-confirmed
cohort. Ann Clin Transl Neurol. 7:2150–2160. 2020.PubMed/NCBI View Article : Google Scholar
|
|
235
|
Toledo JB, Gopal P, Raible K, Irwin DJ,
Brettschneider J, Sedor S, Waits K, Boluda S, Grossman M, Van
Deerlin VM, et al: Pathological ?-synuclein distribution in
subjects with coincident Alzheimer's and Lewy body pathology. Acta
Neuropathol. 131:393–409. 2016.PubMed/NCBI View Article : Google Scholar
|
|
236
|
Howard E, Irwin DJ, Rascovsky K, Nevler N,
Shellikeri S, Tropea TF, Spindler M, Deik A, Chen-Plotkin A,
Siderowf A, et al: Cognitive Profile and Markers of Alzheimer
disease-type pathology in patients with lewy body dementias.
Neurology. 96:e1855–e1864. 2021.PubMed/NCBI View Article : Google Scholar
|
|
237
|
Tan MMX, Lawton MA, Jabbari E, Reynolds
RH, Iwaki H, Blauwendraat C, Kanavou S, Pollard MI, Hubbard L,
Malek N, et al: Genome-Wide Association Studies of Cognitive and
Motor Progression in Parkinson's disease. Mov Disord. 36:424–433.
2021.PubMed/NCBI View Article : Google Scholar
|
|
238
|
Hopfner F, Mueller SH, Szymczak S, Junge
O, Tittmann L, May S, Lohmann K, Grallert H, Lieb W, Strauch K, et
al: Rare variants in specific lysosomal genes are associated with
Parkinson's disease. Mov Disord. 35:1245–1248. 2020.PubMed/NCBI View Article : Google Scholar
|
|
239
|
Setó-Salvia N, Pagonabarraga J, Houlden H,
Pascual-Sedano B, Dols-Icardo O, Tucci A, Paisán-Ruiz C, Campolongo
A, Antón-Aguirre S, Martín I, et al: Glucocerebrosidase mutations
confer a greater risk of dementia during Parkinson's disease
course. Mov Disord. 27:393–399. 2012.PubMed/NCBI View Article : Google Scholar
|
|
240
|
Chia R, Sabir MS, Bandres-Ciga S,
Saez-Atienzar S, Reynolds RH, Gustavsson E, Walton RL, Ahmed S,
Viollet C, Ding J, et al: Genome sequencing analysis identifies new
loci associated with Lewy body dementia and provides insights into
its genetic architecture. Nat Genet. 53:294–303. 2021.PubMed/NCBI View Article : Google Scholar
|
|
241
|
Jiang Z, Huang Y, Zhang P, Han C, Lu Y, Mo
Z, Zhang Z, Li X, Zhao S, Cai F, et al: Characterization of a
pathogenic variant in GBA for Parkinson's disease with mild
cognitive impairment patients. Mol Brain. 13(102)2020.PubMed/NCBI View Article : Google Scholar
|
|
242
|
Martelle SE, Raffield LM, Palmer ND, Cox
AJ, Freedman BI, Hugenschmidt CE, Williamson JD and Bowden DW:
Dopamine pathway gene variants may modulate cognitive performance
in the DHS-Mind Study. Brain Behav. 6(e0044)2016.PubMed/NCBI View Article : Google Scholar
|
|
243
|
Bäckström D, Granåsen G, Domellöf ME,
Linder J, Jakobson Mo S, Riklund K, Zetterberg H, Blennow K and
Forsgren L: Early predictors of mortality in parkinsonism and
Parkinson disease: A population-based study. Neurology.
91:e2045–e2056. 2018.PubMed/NCBI View Article : Google Scholar
|
|
244
|
Aarsland D, Perry R, Brown A, Larsen JP
and Ballard C: Neuropathology of dementia in Parkinson's disease: A
prospective, community-based study. Ann Neurol. 58:773–776.
2005.PubMed/NCBI View Article : Google Scholar
|
|
245
|
Colosimo C and Riario Sforza A: Diagnostic
imaging of degenerative diseases of the extrapyramidal system.
Radiol Med. 106 (3 Suppl 1):S19–S23. 2003.PubMed/NCBI(In Italian).
|
|
246
|
Wakabayashi K, Takahashi H, Takeda S,
Ohama E and Ikuta F: Parkinson's disease: The presence of Lewy
bodies in Auerbach's and Meissner's plexuses. Acta Neuropathol.
76:217–221. 1988.PubMed/NCBI View Article : Google Scholar
|
|
247
|
Braak H, Del Tredici K, Rub U, de Vos RA,
Jansen Steur EN and Braak E: Staging of brain pathology related to
sporadic Parkinson's disease. Neurobiol Aging. 24:197–211.
2003.PubMed/NCBI View Article : Google Scholar
|
|
248
|
Kosaka K: Diffuse Lewy body disease in
Japan. J Neurol. 237:197–204. 1990.PubMed/NCBI View Article : Google Scholar
|
|
249
|
Gibb WR: Idiopathic Parkinson's disease
and the Lewy body disorders. Neuropathol Appl Neurobiol.
12:223–234. 1986.PubMed/NCBI View Article : Google Scholar
|
|
250
|
Braak H, Rub U, Jansen Steur EN, Del
Tredici K and de Vos RA: Cognitive status correlates with
neuropathologic stage in Parkinson disease. Neurology.
64:1404–1410. 2005.PubMed/NCBI View Article : Google Scholar
|
|
251
|
Horimoto Y, Matsumoto M, Akatsu H, Ikari
H, Kojima K, Yamamoto T, Otsuka Y, Ojika K, Ueda R and Kosaka K:
Autonomic dysfunctions in dementia with Lewy bodies. J Neurol.
250:530–533. 2003.PubMed/NCBI View Article : Google Scholar
|
|
252
|
Orimo S, Amino T, Itoh Y, Takahashi A,
Kojo T, Uchihara T, Tsuchiya K, Mori F, Wakabayashi K and Takahashi
H: Cardiac sympathetic denervation precedes neuronal loss in the
sympathetic ganglia in Lewy body disease. Acta Neuropathol.
109:583–588. 2005.PubMed/NCBI View Article : Google Scholar
|
|
253
|
Larner AJ, Mathias CJ and Rossor MN:
Autonomic failure preceding dementia with Lewy bodies. J Neurol.
247:229–231. 2000.PubMed/NCBI View Article : Google Scholar
|
|
254
|
Mehrabian S, Duron E, Labouree F, Rollot
F, Bune A, Traykov L and Hanon O: Relationship between orthostatic
hypotension and cognitive impairment in the elderly. J Neurol Sci.
299:45–48. 2010.PubMed/NCBI View Article : Google Scholar
|
|
255
|
Moretti R, Torre P, Antonello RM,
Manganaro D, Vilotti C and Pizzolato G: Risk factors for vascular
dementia: Hypotension as a key point. Vasc Health Risk Manag.
4:395–402. 2008.PubMed/NCBI View Article : Google Scholar
|
|
256
|
Coon EA, Cutsforth-Gregory JK and
Benarroch EE: Neuropathology of autonomic dysfunction in
synucleinopathies. Mov Disord. 33:349–358. 2018.PubMed/NCBI View Article : Google Scholar
|
|
257
|
De Pablo-Fernandez E, Courtney R, Warner
TT and Holton JL: A histologic study of the circadian system in
Parkinson disease, multiple system atrophy, and progressive
supranuclear Palsy. JAMA Neurol. 75:1008–1012. 2018.PubMed/NCBI View Article : Google Scholar
|
|
258
|
Sharabi Y and Goldstein DS: Mechanisms of
orthostatic hypotension and supine hypertension in Parkinson
disease. J Neurol Sci. 310:123–128. 2011.PubMed/NCBI View Article : Google Scholar
|
|
259
|
Treglia G, Cason E, Gabellini A, Giordano
A and Fagioli G: Recent developments in innervation imaging using
iodine-123-metaiodobenzylguanidine scintigraphy in Lewy body
diseases. Neurol Sci. 31:417–422. 2010.PubMed/NCBI View Article : Google Scholar
|
|
260
|
Iwanaga K, Wakabayashi K, Yoshimoto M,
Tomita I, Satoh H, Takashima H, Satoh A, Seto M, Tsujihata M and
Takahashi H: Lewy body-type degeneration in cardiac plexus in
Parkinson's and incidental Lewy body diseases. Neurology.
52:1269–1271. 1999.PubMed/NCBI View Article : Google Scholar
|
|
261
|
Goldstein DS, Eisenhofer G, Stull R, Folio
CJ, Keiser HR and Kopin IJ: Plasma dihydroxyphenylglycol and the
intraneuronal disposition of norepinephrine in humans. J Clin
Invest. 81:213–220. 1988.PubMed/NCBI View Article : Google Scholar
|
|
262
|
Gibbons CH, Schmidt P, Biaggioni I,
Frazier-Mills C, Freeman R, Isaacson S, Karabin B, Kuritzky L, Lew
M, Low P, et al: The recommendations of a consensus panel for the
screening, diagnosis, and treatment of neurogenic orthostatic
hypotension and associated supine hypertension. J Neurol.
264:1567–1582. 2017.PubMed/NCBI View Article : Google Scholar
|
|
263
|
Tipre DN and Goldstein DS: Cardiac and
extracardiac sympathetic denervation in Parkinson's disease with
orthostatic hypotension and in pure autonomic failure. J Nucl Med.
46:1775–1781. 2005.PubMed/NCBI
|
|
264
|
Noack C, Schroeder C, Heusser K and Lipp
A: Cardiovascular effects of levodopa in Parkinson's disease.
Parkinsonism Relat Disord. 20:815–818. 2014.PubMed/NCBI View Article : Google Scholar
|
|
265
|
Hastings MH, Maywood ES and Brancaccio M:
Generation of circadian rhythms in the suprachiasmatic nucleus. Nat
Rev Neurosci. 19:453–469. 2018.PubMed/NCBI View Article : Google Scholar
|
|
266
|
Videnovic A, Noble C, Reid KJ, Peng J,
Turek FW, Marconi A, Rademaker AW, Simuni T, Zadikoff C and Zee PC:
Circadian melatonin rhythm and excessive daytime sleepiness in
Parkinson disease. JAMA Neurol. 71:463–469. 2014.PubMed/NCBI View Article : Google Scholar
|
|
267
|
Espay AJ, LeWitt PA and Kaufmann H:
Norepinephrine deficiency in Parkinson's disease: The case for
noradrenergic enhancement. Mov Disord. 29:1710–1719.
2014.PubMed/NCBI View Article : Google Scholar
|
|
268
|
Kato S, Oda M, Hayashi H, Shimizu T,
Hayashi M, Kawata A and Tanabe H: Decrease of medullary
catecholaminergic neurons in multiple system atrophy and
Parkinson's disease and their preservation in amyotrophic lateral
sclerosis. J Neurol Sci. 132:216–221. 1995.PubMed/NCBI View Article : Google Scholar
|
|
269
|
Dayan E, Sklerov M and Browner N:
Disrupted hypothalamic functional connectivity in patients with
Parkinson's disease and autonomic dysfunction. Neurology.
90:e2051–e2058. 2018.PubMed/NCBI View Article : Google Scholar
|
|
270
|
Papapetropoulos S and Mash DC: Insular
pathology in Parkinson's disease patients with orthostatic
hypotension. Parkinsonism Relat Disord. 13:308–311. 2007.PubMed/NCBI View Article : Google Scholar
|
|
271
|
McAleese KE, Alafuzoff I, Charidimou A, De
Reuck J, Grinberg LT, Hainsworth AH, Hortobagyi T, Ince P,
Jellinger K, Gao J, et al: Post-mortem assessment in vascular
dementia: Advances and aspirations. BMC Med. 14(129)2016.PubMed/NCBI View Article : Google Scholar
|
|
272
|
Prins ND and Scheltens P: White matter
hyperintensities, cognitive impairment and dementia: An update. Nat
Rev Neurol. 11:157–165. 2015.PubMed/NCBI View Article : Google Scholar
|
|
273
|
Dadar M, Maranzano J, Ducharme S and
Collins DL: Alzheimer's Disease Neuroimaging Initiative. White
matter in different regions evolves differently during progression
to dementia. Neurobiol Aging. 76:71–79. 2019.PubMed/NCBI View Article : Google Scholar
|
|
274
|
Gunning-Dixon FM and Raz N: The cognitive
correlates of white matter abnormalities in normal aging: A
quantitative review. Neuropsychology. 14:224–232. 2000.PubMed/NCBI View Article : Google Scholar
|
|
275
|
Bohnen NI and Albin RL: White matter
lesions in Parkinson disease. Nat Rev Neurol. 7:229–236.
2011.PubMed/NCBI View Article : Google Scholar
|
|
276
|
Lee SJ, Kim JS, Yoo JY, Song IU, Kim BS,
Jung SL, Yang DW, Kim YI, Jeong DS and Lee KS: Influence of white
matter hyperintensities on the cognition of patients with Parkinson
disease. Alzheimer Assoc Disord. 24:227–233. 2010.PubMed/NCBI View Article : Google Scholar
|
|
277
|
Chesebro AG, Melgarejo JD, Leendertz R,
Igwe KC, Lao PJ, Laing KK, Rizvi B, Budge M, Meier IB, Calmon G, et
al: White matter hyperintensities mediate the association of
nocturnal blood pressure with cognition. Neurology. 94:e1803–e1810.
2020.PubMed/NCBI View Article : Google Scholar
|
|
278
|
Li Q, Yang Y, Reis C, Tao T, Li W, Li X
and Zhang JH: Cerebral small vessel disease. Cell Transpl.
27:1711–1722. 2018.PubMed/NCBI View Article : Google Scholar
|
|
279
|
Iadecola C and Gottesman RF: Neurovascular
and cognitive dysfunction in hypertension. Circ Res. 124:1025–1044.
2019.PubMed/NCBI View Article : Google Scholar
|
|
280
|
Kaufmann H and Palma JA: White matter
hyperintensities in the Synucleinopathies: Orthostatic Hypotension,
Supine Hypertension, or Both? Mov Disord Clin Pract. 7:595–598.
2020.PubMed/NCBI View Article : Google Scholar
|
|
281
|
Palma JA and Kaufmann H: Epidemiology,
Diagnosis, and Management of Neurogenic Orthostatic Hypotension.
Mov Disord Clin Pract. 4:298–308. 2017.PubMed/NCBI View Article : Google Scholar
|
|
282
|
Biaggioni I, Robertson D, Krantz S, Jones
M and Haile V: The anemia of primary autonomic failure and its
reversal with recombinant erythropoietin. Ann Intern Med.
121:181–186. 1994.PubMed/NCBI View Article : Google Scholar
|
|
283
|
Perera R, Isola L and Kaufmann H: Effect
of recombinant erythropoietin on anemia and Orthostatic Hypotension
in primary autonomic failure. Clin Auton Res. 5:211–213.
1995.PubMed/NCBI View Article : Google Scholar
|
|
284
|
May M and Jordan J: The osmopressor
response to water drinking. Am J Physiol Regul Integr Comp Physiol.
300:R40–R46. 2011.PubMed/NCBI View Article : Google Scholar
|
|
285
|
Shibao C, Gamboa A, Diedrich A, Dossett C,
Choi L, Farley G and Biaggioni I: Acarbose, an alpha-glucosidase
inhibitor, attenuates postprandial hypotension in autonomic
failure. Hypertension. 50:54–61. 2007.PubMed/NCBI View Article : Google Scholar
|
|
286
|
Freeman R: Clinical practice. Neurogenic
orthostatic hypotension. N Engl J Med. 358:615–624. 2008.PubMed/NCBI View Article : Google Scholar
|
|
287
|
Low DA, Vichayanrat E, Iodice V and
Mathias CJ: Exercise hemodynamics in Parkinson's disease and
autonomic dysfunction. Parkinson's disease Relat Disord.
20:549–553. 2014.PubMed/NCBI View Article : Google Scholar
|
|
288
|
Kooner JS, Raimbach S, Watson L, Bannister
R, Peart S and Mathias CJ: Relationship between splanchnic
vasodilation and postprandial hypotension in patients with primary
autonomic failure. J Hypertens. (Suppl 7):S40–S41. 1989.PubMed/NCBI View Article : Google Scholar
|
|
289
|
Mata IF, Leverenz JB, Weintraub D,
Trojanowski JQ, Hurtig HI, Van Deerlin VM, Ritz B, Rausch R, Rhodes
SL, Factor SA, et al: APOE, MAPT, and SNCA genes and cognitive
performance in Parkinson disease. JAMA Neurol. 71:1405–1412.
2014.PubMed/NCBI View Article : Google Scholar
|
|
290
|
Krediet CT, van Lieshout JJ, Bogert LW,
Immink RV, Kim YS and Wieling W: Leg crossing improves orthostatic
tolerance in healthy subjects: A placebo-controlled crossover
study. Am J Physiol Heart Circ Physiol. 291:H1768–H1772.
2006.PubMed/NCBI View Article : Google Scholar
|
|
291
|
Logan A, Freeman J, Pooler J, Kent B, Gunn
H, Billings S, Cork E and Marsden J: Effectiveness of
non-pharmacological interventions to treat orthostatic hypotension
in elderly people and people with a neurological condition: A
systematic review. JBI Evid Synth. 18:2556–2617. 2020.PubMed/NCBI View Article : Google Scholar
|
|
292
|
Protheroe CL, Dikareva A, Menon C and
Claydon VE: Are compression stockings an effective treatment for
orthostatic presyncope? PLoS One. 6(e28193)2011.PubMed/NCBI View Article : Google Scholar
|
|
293
|
Fanciulli A, Leys F, Falup-Pecurariu C,
Thijs R and Wenning GK: Management of Orthostatic Hypotension in
Parkinson's Disease. J Parkinsons Dis. 10(S1):S57–S64.
2020.PubMed/NCBI View Article : Google Scholar
|
|
294
|
Chobanian AV, Volicer L, Tifft CP, Gavras
H, Liang CS and Faxon D: Mineralocorticoid-induced hypertension in
patients with Orthostatic Hypotension. N Engl J Med. 301:68–73.
1979.PubMed/NCBI View Article : Google Scholar
|
|
295
|
Grijalva CG, Biaggioni I, Griffin MR and
Shibao CA: Fludrocortisone is associated with a higher risk of
all-cause hospitalizations compared with midodrine in patients with
Orthostatic Hypotension. J Am Heart Assoc.
6(e006848)2017.PubMed/NCBI View Article : Google Scholar
|
|
296
|
Norcliffe-Kaufmann L, Axelrod FB and
Kaufmann H: Developmental abnormalities, blood pressure variability
and renal disease in Riley Day syndrome. J Hum Hypertens. 27:51–55.
2013.PubMed/NCBI View Article : Google Scholar
|
|
297
|
Campbell IW, Ewing DJ and Clarke BF:
9-Alpha-fluorohydrocortisone in the treatment of postural
hypotension in diabetic autonomic neuropathy. Diabetes. 24:381–384.
1975.PubMed/NCBI View Article : Google Scholar
|
|
298
|
Decaux G: Fludrocortisone in Orthostatic
Hypotension. N Engl J Med. 301:1121–1122. 1979.PubMed/NCBI
|
|
299
|
Van Lieshout JJ, Ten Harkel AD and Wieling
W: Fludrocortisone and sleeping in the head-up position limit the
postural decrease in cardiac output in autonomic failure. Clin
Auton Res. 10:35–42. 2000.PubMed/NCBI View Article : Google Scholar
|
|
300
|
Watt SJ, Tooke JE, Perkins CM and Lee MR:
The treatment of idiopathic Orthostatic Hypotension: A combined
fludrocortisone and flurbiprofen regime. Q J Med. 50:205–212.
1981.PubMed/NCBI
|
|
301
|
Low PA and Singer W: Management of
neurogenic orthostatic hypotension: An update. Lancet Neurol.
7:451–458. 2008.PubMed/NCBI View Article : Google Scholar
|
|
302
|
Low PA, Gilden JL, Freeman R, Sheng KN and
McElligott MA: Efficacy of midodrine vs placebo in neurogenic
orthostatic hypotension. A randomized, double-blind multicenter
study. Midodrine Study Group. JAMA. 277:1046–1051. 1997.PubMed/NCBI
|
|
303
|
Wright RA, Kaufmann HC, Perera R,
Opfer-Gehrking TL, McElligott MA, Sheng KN and Low PA: A
double-blind, dose-response study of midodrine in neurogenic
orthostatic hypotension. Neurology. 51:120–124. 1998.PubMed/NCBI View Article : Google Scholar
|
|
304
|
McTavish D and Goa KL: Midodrine. A review
of its pharmacological properties and therapeutic use in
orthostatic hypotension and secondary hypotensive disorders. Drugs.
38:757–777. 1989.PubMed/NCBI View Article : Google Scholar
|
|
305
|
Kaufmann H, Norcliffe-Kaufmann L and Palma
JA: Droxidopa in neurogenic orthostatic hypotension. Expert Rev
Cardiovasc Ther. 13:875–891. 2015.PubMed/NCBI View Article : Google Scholar
|
|
306
|
Goodman BP, Claassen D and Mehdirad A:
Adjusting droxidopa for neurogenic orthostatic hypotension in a
patient with Parkinson disease. Clin Auton Res. 27 (Suppl
1):S17–S19. 2017.PubMed/NCBI View Article : Google Scholar
|
|
307
|
Gupta F, Karabin B and Mehdirad A:
Titrating droxidopa to maximize symptomatic benefit in a patient
with Parkinson disease and neurogenic orthostatic hypotension. Clin
Auton Res. 27:S15–S16. 2017.PubMed/NCBI View Article : Google Scholar
|
|
308
|
Cheshire WP: Chemical pharmacotherapy for
the treatment of orthostatic hypotension. Expert Opin Pharmacother.
20:187–199. 2019.PubMed/NCBI View Article : Google Scholar
|
|
309
|
Okamoto LE, Shibao C, Gamboa A, Choi L,
Diedrich A, Raj SR, Black BK, Robertson D and Biaggioni I:
Synergistic effect of norepinephrine transporter blockade and α-2
antagonism on blood pressure in autonomic failure. Hypertension.
59:650–656. 2012.PubMed/NCBI View Article : Google Scholar
|
|
310
|
Ramirez CE, Okamoto LE, Arnold AC, Gamboa
A, Diedrich A, Choi L, Raj SR, Robertson D, Biaggioni I and Shibao
CA: Efficacy of atomoxetine versus midodrine for the treatment of
orthostatic hypotension in autonomic failure. Hypertension.
64:1235–1240. 2014.PubMed/NCBI View Article : Google Scholar
|
|
311
|
Shibao CA, Palma JA, Celedonio JE,
Martinez J, Kaufmann H and Biaggioni I: Predictors of the pressor
response to the norepinephrine transporter inhibitor, atomoxetine.
neurogenic orthostatic hypotension. Hypertension. 78:525–531.
2021.PubMed/NCBI View Article : Google Scholar
|
|
312
|
Kim JS, Ryu DW, Oh JH, Lee YH, Park SJ,
Jeon K, Lee JY, Ho SH, So J, Im JH and Lee KS: Cardiovascular
autonomic dysfunction in patients with drug-induced parkinsonism. J
Clin Neurol. 13:15–20. 2017.PubMed/NCBI View Article : Google Scholar
|
|
313
|
Singer W, Sandroni P, Opfer-Gehrking TL,
Suarez GA, Klein CM, Hines S, O'Brien PC, Slezak J and Low PA:
Pyridostigmine treatment trial in neurogenic orthostatic
hypotension. Arch Neurol. 63:513–518. 2006.PubMed/NCBI View Article : Google Scholar
|
|
314
|
Byun JI, Moon J, Kim DY, Shin H, Sunwoo
JS, Lim JA, Kim TJ, Lee WJ, Lee HS, Jun J, et al: Efficacy of
single or combined midodrine and pyridostigmine in orthostatic
hypotension. Neurology. 89:1078–1086. 2017.PubMed/NCBI View Article : Google Scholar
|
|
315
|
Eschlböck S, Wenning G and Fanciulli A:
Evidence-based treatment of neurogenic orthostatic hypotension and
related symptoms. J Neural Transm (Vienna). 124:1567–1605.
2017.PubMed/NCBI View Article : Google Scholar
|
|
316
|
Shibao C, Okamoto L and Biaggioni I:
Pharmacotherapy of autonomic failure. Pharmacol Ther. 134:279–286.
2012.PubMed/NCBI View Article : Google Scholar
|
|
317
|
Mazza A, Ravenni R, Antonini A, Casiglia
E, Rubello D and Pauletto P: Arterial hypertension, a tricky side
of Parkinson's disease: Physiopathology and therapeutic features.
Neurol Sci. 34:621–627. 2013.PubMed/NCBI View Article : Google Scholar
|
|
318
|
Zeng G and Quon MJ: Insulin-stimulated
production of nitric oxide is inhibited by wortmannin. Direct
measurement in vascular endothelial cells. J Clin Invest.
98:894–898. 1996.PubMed/NCBI View Article : Google Scholar
|
|
319
|
Okamoto LE, Celedonio JE, Smith EC, Gamboa
A, Shibao CA, Diedrich A, Paranjape SY, Black BK, Muldowney JAS
III, Peltier AC, et al: Local Passive Heat for the Treatment of
Hypertension in Autonomic Failure. J Am Heart Assoc.
10(e018979)2021.PubMed/NCBI View Article : Google Scholar
|
|
320
|
Okamoto LE, Celedonio JE, Smith EC,
Paranjape SY, Black BK, Wahba A, Park JW, Shibao CA, Diedrich A and
Biaggioni I: Continuous positive airway pressure for the treatment
of supine hypertension and orthostatic hypotension in autonomic
failure. Hypertension. 80:650–658. 2023.PubMed/NCBI View Article : Google Scholar
|
|
321
|
Arnold AC, Okamoto LE, Gamboa A, Black BK,
Raj SR, Elijovich F, Robertson D, Shibao CA and Biaggioni I:
Mineralocorticoid receptor activation contributes to the supine
hypertension of autonomic failure. Hypertension. 67:424–429.
2016.PubMed/NCBI View Article : Google Scholar
|
|
322
|
Jordan J, Fanciulli A, Tank J,
Calandra-Buonaura G, Cheshire WP, Cortelli P, Eschlboeck S, Grassi
G, Hilz MJ, Kaufmann H, et al: Management of supine hypertension in
patients with neurogenic orthostatic hypotension: Scientific
statement of the American Autonomic Society, European Federation of
Autonomic Societies, and the European Society of Hypertension. J
Hypertens. 37:1541–1546. 2019.PubMed/NCBI View Article : Google Scholar
|
|
323
|
Gomperts SN: Lewy body dementias: Dementia
with Lewy bodies and Parkinson disease dementia. Continuum (Minneap
Minn). 22 (2 Dementia):435–463. 2016.PubMed/NCBI View Article : Google Scholar
|
|
324
|
Walker Z, Possin KL, Boeve BF and Aarsland
D: Lewy body dementias. Lancet. 386:1683–1697. 2015.PubMed/NCBI View Article : Google Scholar
|
|
325
|
Goodnick PJ and Goldstein BJ: Selective
serotonin reuptake inhibitors in affective disorders-I. Basic
pharmacology. J Psychopharmacol. 12 (3 Suppl B):S5–S20.
1998.PubMed/NCBI View Article : Google Scholar
|
|
326
|
Owens MJ, Morgan WN, Plott SJ and Nemeroff
CB: Neurotransmitter receptor and transporter binding profile of
antidepressants and their metabolites. J Pharmacol Exp Ther.
283:1305–1322. 1997.PubMed/NCBI
|
|
327
|
Wagg A, Dale M, Tretter R, Stow B and
Compion G: Randomised, multicentre, placebo-controlled,
double-blind crossover study investigating the effect of
solifenacin and oxybutynin in elderly people with mild cognitive
impairment: the SENIOR study. Eur Urol. 64:74–81. 2013.PubMed/NCBI View Article : Google Scholar
|
|
328
|
Peroutka SJ and Snyder SH: Antiemetics:
Neurotransmitter receptor binding predicts therapeutic actions.
Lancet. 1:658–659. 1982.PubMed/NCBI View Article : Google Scholar
|
|
329
|
Wallace DM, Wohlgemuth WK, Trotti LM,
Amara AW, Malaty IA, Factor SA, Nallu S, Wittine L and Hauser RA:
Practical evaluation and management of insomnia in Parkinson's
Disease: A review. Mov Disord Clin Pract. 7:250–266.
2020.PubMed/NCBI View Article : Google Scholar
|
|
330
|
Orgeta V, McDonald KR, Poliakoff E, Hindle
JV, Clare L and Leroi I: Cognitive training interventions for
dementia and mild cognitive impairment in Parkinson's Disease.
Cochrane Database Syst Rev. 2(CD011961)2020.PubMed/NCBI View Article : Google Scholar
|
|
331
|
da Silva FC, Iop RDR, de Oliveira LC, Boll
AM, de Alvarenga JGS, Gutierres Filho PJB, de Melo LMAB, Xavier AJ
and da Silva R: Effects of physical exercise programs on cognitive
function in Parkinson's Disease patients: A systematic review of
randomized controlled trials of the last 10 years. PLoS One.
13(e0193113)2018.PubMed/NCBI View Article : Google Scholar
|
|
332
|
Picelli A, Varalta V, Melotti C, Zatezalo
V, Fonte C, Amato S, Saltuari L, Santamato A, Fiore P and Smania N:
Effects of treadmill training on cognitive and motor features of
patients with mild to moderate Parkinson's Disease: A pilot,
single-blind, randomized controlled trial. Funct Neurol. 31:25–31.
2016.PubMed/NCBI View Article : Google Scholar
|
|
333
|
Trung J, Hanganu A, Jobert S, Degroot C,
Mejia-Constain B, Kibreab M, Bruneau MA, Lafontaine AL, Strafella A
and Monchi O: Transcranial magnetic stimulation improves cognition
over time in Parkinson's Disease. Parkinsonism Relat Disord.
66:3–8. 2019.PubMed/NCBI View Article : Google Scholar
|
|
334
|
Doruk D, Gray Z, Bravo GL, Pascual-Leone A
and Fregni F: Effects of tDCS on executive function in Parkinson's
Disease. Neurosci Lett. 582:27–31. 2014.PubMed/NCBI View Article : Google Scholar
|
|
335
|
Manenti R, Cotelli MS, Cobelli C, Gobbi E,
Brambilla M, Rusich D, Alberici A, Padovani A, Borroni B and
Cotelli M: Transcranial direct current stimulation combined with
cognitive training for the treatment of Parkinson Disease: A
randomized, placebo-controlled study. Brain Stimulat. 11:1251–1262.
2018.PubMed/NCBI View Article : Google Scholar
|
|
336
|
Arendt T, Bigl V, Arendt A and Tennstedt
A: Loss of neurons in the nucleus basalis of Meynert in Alzheimer's
disease, paralysis agitans and Korsakoff's Disease. Acta
Neuropathol. 61:101–108. 1983.PubMed/NCBI View Article : Google Scholar
|
|
337
|
Kandiah N, Pai MC, Senanarong V, Looi I,
Ampil E, Park KW, Karanam AK and Christopher S: Rivastigmine: The
advantages of dual inhibition of acetylcholinesterase and
butyrylcholinesterase and its role in subcortical vascular dementia
and Parkinson's Disease dementia. Clin Interv Aging. 12:697–707.
2017.PubMed/NCBI View Article : Google Scholar
|
|
338
|
Devos D, Moreau C, Maltête D, Lefaucheur
R, Kreisler A, Eusebio A, Defer G, Ouk T, Azulay JP, Krystkowiak P,
et al: Rivastigmine in apathetic but dementia and depression-free
patients with Parkinson's Disease: A double-blind,
placebo-controlled, randomised clinical trial. J Neurol Neurosurg
Psychiatry. 85:668–674. 2014.PubMed/NCBI View Article : Google Scholar
|
|
339
|
Viramo P, Luukinen H, Koski K, Laippala P,
Sulkava R and Kivela SL: Orthostatic hypotension and cognitive
decline in older people. J Am Geriatr Soc. 47:600–604.
1999.PubMed/NCBI View Article : Google Scholar
|
|
340
|
Kuo HK, Sorond F, Iloputaife I, Gagno M,
Milberg W and Lipsitz LA: Effect of blood pressure on cognitive
functions in elderly persons. J Gerontol Biol Sci Med Sci.
59:1191–1194. 2004.PubMed/NCBI View Article : Google Scholar
|
|
341
|
Allcock LM, Kenny RA and Burn DJ: Clinical
phenotype of subjects with Parkinson's disease and orthostatic
hypotension: Autonomic symptom and demographic comparison. Mov
Disord. 21:1851–1855. 2006.PubMed/NCBI View Article : Google Scholar
|
|
342
|
Yap PL, Niti M, Yap KB and Ng TP:
Orthostatic hypotension, hypotension and cognitive status: Early
comorbid markers of primary dementia? Dement Geriatr Cogn Disord.
26:239–246. 2008.PubMed/NCBI View Article : Google Scholar
|
|
343
|
Bendini C, Angelini A, Salsi F, Finelli
ME, Martini E, Neviani F, Mussi C and Neri M: Relation of
neurocardiovascular instability to cognitive, emotional and
functional domains. Arch Gerontol Geriatr. 44 (Suppl 1):S69–S74.
2007.PubMed/NCBI View Article : Google Scholar
|
|
344
|
Rose KM, Couper D, Eigenbrodt ML, Mosley
TH, Sharrett AR and Gottesman RF: Orthostatic hypotension and
cognitive function: The Atherosclerosis Risk in Communities Study.
Neuroepidemiology. 34:1–7. 2010.PubMed/NCBI View Article : Google Scholar
|
|
345
|
Hauser RA, Heritier S, Rowse GJ, Hewitt LA
and Isaacson SH: Droxidopa and reduced falls in a trial of
Parkinson disease patients with neurogenic orthostatic hypotension.
Clin Neuropharmacol. 39:220–226. 2016.PubMed/NCBI View Article : Google Scholar
|
|
346
|
Brignole M, Moya A, de Lange FJ, Deharo
JC, Elliott PM, Fanciulli A, Fedorowski A, Furlan R, Kenny RA,
Martín A, et al: 2018 ESC Guidelines for the diagnosis and
management of syncope. Eur Heart J. 39:1883–1948. 2018.PubMed/NCBI View Article : Google Scholar
|
|
347
|
Shannon J, Jordan J, Costa F, Robertson RM
and Biaggioni I: The hypertension of autonomic failure and its
treatment. Hypertension. 30:1062–1067. 1997.PubMed/NCBI View Article : Google Scholar
|
|
348
|
Arnold AC, Okamoto LE, Gamboa A, Shibao C,
Raj SR, Robertson D and Biaggioni I: Angiotensin II, independent of
plasma renin activity, contributes to the hypertension of autonomic
failure. Hypertension. 61:701–706. 2013.PubMed/NCBI View Article : Google Scholar
|
|
349
|
Jordan J, Shannon JR, Pohar B, Paranjape
SY, Robertson D, Robertson RM and Biaggioni I: Contrasting effects
of vasodilators on blood pressure and sodium balance in the
hypertension of autonomic failure. J Am Soc Nephrol. 10:35–42.
1999.PubMed/NCBI View Article : Google Scholar
|
|
350
|
Dubois B, Tolosa E, Katzenschlager R, Emre
M, Lees AJ, Schumann G, Pourcher E, Gray J, Thomas G, Swartz J, et
al: Donepezil in Parkinson's disease dementia: A randomized,
double-blind efficacy and safety study. Mov Disord. 27:1230–1238.
2012.PubMed/NCBI View Article : Google Scholar
|
|
351
|
Wang HF, Yu JT, Tang SW, Jiang T, Tan CC,
Meng XF, Wang C, Tan MS and Tan L: Efficacy and safety of
cholinesterase inhibitors and memantine in cognitive impairment in
Parkinson's disease, Parkinson's disease dementia, and dementia
with Lewy bodies: Systematic review with meta-analysis and trial
sequential analysis. J Neurol Neurosurg Psychiatry. 86:135–143.
2015.PubMed/NCBI View Article : Google Scholar
|
|
352
|
Litvan I, Goldman JG, Troster AI, Schmand
BA, Weintraub D, Petersen RC, Mollenhauer B, Adler CH, Marder K,
Williams-Gray CH, et al: Diagnostic criteria for mild cognitive
impairment in Parkinson's disease: Movement disorder society task
force guidelines. Mov Disord. 27:349–356. 2012.PubMed/NCBI View Article : Google Scholar
|
|
353
|
Emre M, Tsolaki M, Bonuccelli U, Destee A,
Tolosa E, Kutzelnigg A, Ceballos-Baumann A, Zdravkovic S, Bladström
A and Jones R: 11018 Study Investigators. Memantine for patients
with Parkinson's disease dementia or dementia with Lewy bodies: A
randomised, double-blind, placebo-controlled trial. Lancet Neuro.
9:969–977. 2010.PubMed/NCBI View Article : Google Scholar
|