Introduction
Lung carcinoma is the most common cancer in the
world and the leading cause of cancer-related mortality, with over
one million cases annually (1).
Lung carcinomas are usually classified as small-cell lung
carcinomas (SCLCs) or non-small-cell lung carcinomas (NSCLCs).
NSCLCs are histopathologically and clinically distinct from SCLCs
and are further subcategorized as adenocarcinomas (ACs), squamous
cell carcinomas or large-cell carcinomas, of which ACs are the
predominant form (2).
Several molecular changes are characteristic of lung
AC. These include mutations in the tyrosine kinase domain of the
epidermal growth factor receptor (EGFR), v-Ki-ras2 Kirsten
rat sarcoma viral oncogene homolog (KRAS) (3,4),
tumor protein 53 (TP53) (5), serine/threonine kinase 11
(STK11) (6) and
cyclin-dependent kinase inhibitor 2A (CDKN2A) (7) genes. Among them, somatic-activating
EGFR mutations, particularly deletions in exon 19 and L858R
point mutations in exon 21, may activate the gp130/JAK/STAT3
pathway by means of interleukin 6 (IL-6) upregulation in
primary human lung AC, thereby promoting cell cycle progression,
cell growth and tumorigenesis (8).
Further studies have revealed that thyroid transcription factor-1
(TTF-1) expression is positively associated with EGFR
mutations in lung AC (9). These
results indicate that transcriptional regulation is a fundamental
process in lung AC development.
However, high-throughput functional analyses of
multiple transcription factors (TFs) and their target genes in lung
AC are rare. Therefore, the objective of this study was to identify
potential transcription regulation correlations between TFs and
differentially expressed genes (DEGs) in lung AC using microarray
data and transcriptional network analysis. In addition, the
underlying molecular mechanisms were explored by KEGG pathway
enrichment.
Materials and methods
Affymetrix microarray data analysis
The transcription profiles of human AC GSE2514
(10) were obtained from a public
functional genomics data repository GEO (http://www.ncbi.nlm.nih.gov/geo/) and are based on the
Affymetrix GPL8300 platform data (Affymetrix Human Genome U95
Version 2 Array) (11). Only 20 AC
chips and 19 control chips were useable. Each pair of samples (one
derived from cancer cells and the other from normal cells)
represented a single patient with AC.
All patients participating in this study were
enrolled in a local Colorado Multiple Institutional Review Board
(COMIRB)-approved protocol for use of remnant tissue with
anonymization and analysis of specimens and clinical data. Informed
consent was obtained from all the patients. All but one of the
patients had a history of smoking. Patients ranged in age from 45
to 73 years of age. Tumors from five males and five females were
used in the study. All specimens for microarray analysis were
obtained at surgery with nine patients undergoing lobectomy and one
wedge resection. Specimens were examined immediately following
removal from the patient and grossly visible solid tumor tissue was
snap-frozen for RNA extraction. The tumors were all invasive ACs,
but five specimens exhibited evidence of bronchoalveolar
differentiation at the edge of tumor nests. Most tumors were low to
intermediate grade and low stage, although two stage III tumors
were included in the analysis.
The limma method (12) was used to identify DEGs. The
original expression datasets from all conditions were extracted
into expression estimates using the robust multiarray average (RMA)
method (13) with the default
settings implemented in Bioconductor and the linear model was
constructed. Only the DEGs with a fold-change >1.5 and p-value
<0.05 were selected.
Regulatory network analysis for AC
The TRANSFAC database contains data on TFs, their
experimentally demonstrated binding sites and regulated genes
(14). The Transcriptional
Regulatory Element Database (TRED) has been built in response to
increasing requirements for an integrated repository for cis- and
trans-regulatory elements in mammals (15). TRED has curated transcriptional
regulation information, including TF binding motifs and
experimental evidence. The curation is currently focused on the
target genes of 36 cancer-related TF families. A total of 774 pairs
of regulatory relationships between 219 TFs and 265 target genes
were collected from TRANSFAC (http://www.gene-regulation.com/pub/databases.html). A
total of 5722 pairs of regulatory relationships between 102 TFs and
2920 target genes were collected from TRED (http://rulai.cshl.edu/TRED/). By integrating the two
regulation datasets, a total of 6328 regulatory relationships
between 276 TFs and 3002 target genes were obtained. To demonstrate
the potential regulatory relationships, the Pearson Correlation
Coefficient (PCC) was calculated for all pair-wise comparisons of
gene expression values between TFs and the DEGs. The regulatory
relationships having an absolute PCC >0.7 were considered to be
significant.
Pathway enrichment and TF-pathway
regulatory network analysis
Kyoto Encyclopedia of Genes and Genomes (KEGG)is a
collection of online databases dealing with genomes, enzymatic
pathways and biological chemicals (16). The PATHWAY database records
networks of molecular interactions in cells, and variants of them
specific to particular organisms (http://www.genome.jp/kegg/). A total of 130 pathways,
involving 2287 genes, were collected from KEGG (updated in July
2011).
DAVID (17), a
high-throughput and integrated data-mining environment, analyzes
gene lists derived from high-throughput genomic experiments. DAVID
was used to identify over-represented KEGG pathways. Pathways with
p<0.05 and a count >2 were considered to be significant.
To further investigate the regulatory relationships
between TFs and significant pathways, we mapped target genes in the
network to pathways and created a regulatory network comprising TFs
and pathways.
Results
Microarray data and regulatory network
analysis
Using the limma package, a total of 915 DEGs with
p<0.05 and fold-change >1.5 were selected. Regulatory
relationships with a PCC >0.7 were considered to be significant.
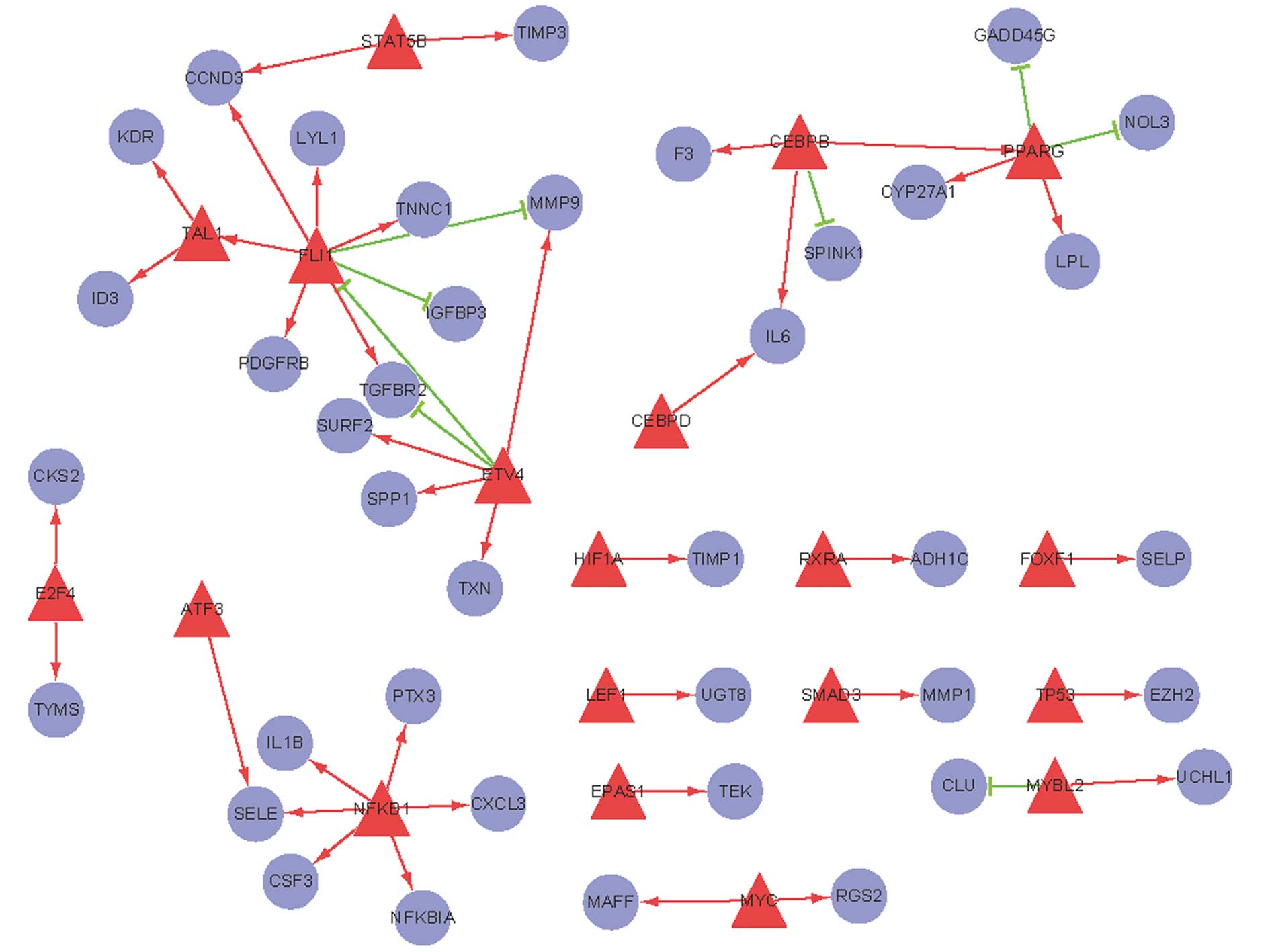
A regulatory network for human AC comprising TFs and their target
genes was constructed (Fig. 1). In
this network, peroxisome proliferator activated receptor-γ
(PPARG), CCAAT/enhancer binding protein [C/EBP], β
(CEBPB), ets variant 4 (ETV4), Friend leukemia virus
integration 1 (FLI1), T-cell acute lymphocytic leukemia 1
(TAL1), and nuclear factor of κ light polypeptide gene enhancer in
B-cells 1 (NFκB1) with higher degrees of interaction formed local
networks, suggesting that these TFs are significant in lung AC.
Among them, it appears that: PPARG can upregulate
lipoprotein lipase (LPL), but downregulate growth arrest and
DNA-damage-inducible, γ (GADD45G) expression; FLI can
upregulate transforming growth factor, β receptor II
(TGFBR2) and cyclin D3 (CCND3) expression;
ETV4 can downregulate TGFBR2 expression, but
stimulate matrix metallopeptidase 9 (MMP9) expression;
NFκB1 can upregulate IL1B expression; CEBPB
can upregulate IL-6 expression; TAL1 could promote
kinase insert domain receptor (KDR) expression.
Significant pathways and TF pathway
regulatory network analysis
Using the KEGG pathways to describe the function of
the regulatory network, several KEGG pathways among the pathways in
the regulatory network were revealed to be enriched, including
pathways in cancer (hsa05200), the PPAR signaling pathway
(hsa03320), cell cycle (hsa04110) and the TGF-β signaling pathway
(hsa04350). The 10 most enriched KEGG pathways are listed in
Table I.
 | Table IPathway significance analysis. |
Table I
Pathway significance analysis.
| Term | Description | Count | p-value | FDR |
|---|
| hsa05200 | Pathways in
cancer | 16 |
1.01e−9 |
1.05e−6 |
| hsa05220 | Chronic myeloid
leukemia | 7 |
9.72e−6 | 0.010121 |
| hsa05216 | Thyroid cancer | 5 |
3.95e−5 | 0.041159 |
| hsa05210 | Colorectal
cancer | 6 |
2.40e−4 | 0.249483 |
| hsa03320 | PPAR signaling
pathway | 5 | 0.001184 | 1.225733 |
| hsa04110 | Cell cycle | 6 | 0.001489 | 1.538652 |
| hsa05222 | Small cell lung
cancer | 5 | 0.002457 | 2.527709 |
| hsa04350 | TGF-β signaling
pathway | 5 | 0.002793 | 2.86916 |
| hsa05219 | Bladder cancer | 4 | 0.002854 | 2.931146 |
| hsa05215 | Prostate
cancer | 5 | 0.003034 | 3.113205 |
To further investigate the regulatory relationships
between TFs and pathways, we mapped DEGs to significant pathways
and obtained a regulatory network comprising TFs and pathways
(Fig. 2). In the network, 9 TFs
regulated 6 pathways. The network indicates that the PPAR pathway
is upregulated by PPARG and CEBPB; the cell cycle is
upregulated by FLI and STAT5B, but downregulated by
PPARG; the TGF-β signaling pathway is upregulated by
FLI and TAL1, but downregulated by ETV4; ETV4
promotes the bladder cancer pathway; NFκB1 promotes the
prostate cancer pathway. Pathways in cancer may be upregulated by
CEBPD and CEBPB.
Discussion
We investigated the comprehensive regulatory network
of lung AC comprising TFs, target genes and their underlying
molecular pathways. In our transcriptosome network, the genes
PPARG, CEBPB, ETV4, FLI1, TAL1,
and NFκB1 are hub nodes. PPARG may promote the PPAR
signaling pathway via upregulation of LPL expression, but
suppress the cell cycle pathway via downregulation of
GADD45G expression; ETV4 can stimulate MMP9
expression to induce the bladder cancer pathway; FLI can
upregulate TGFBR2 expression to activate TGF-β signaling,
and upregulate CCND3 expression to promote the cell cycle
pathway; NFκB1 can upregulate IL1B expression and
initiate the prostate cancer pathway; CEBPB can upregulate
IL-6 expression and promote pathways in cancer; TAL1
could upregulate KDR expression to promote the TGF-β
signaling pathway.
PPARG is a ligand-activated TF, whose activation has
been implicated in the pathology of numerous diseases, including
lung AC. High PPARG expression levels have been detected in lung AC
patients (18) and
PPARG-positivity has been identified more frequently in
well-differentiated AC cases than in moderately and poorly
differentiated ones (19). The
treatment of lung AC cells with PPARG ligands induces a
dose-dependent inhibition of lung AC cell growth, that is, a cell
cycle arrest at G0/G1 (18,20).
GADD45 is a cell cycle-regulated nuclear protein that reaches
maximal levels in the G1 phase of the cycle (21). Through its association with Cdc2,
GADD45 disrupts the interactions of Cdc2 with cyclin B1 and, thus,
may induce G2/M arrest (22).
Previous studies indicate that the activation of PPARG may lead to
apoptosis and growth arrest, at least in part, by inducing the
Oct-1-mediated transcription of GADD45 (23). However, GADD45G methylation is
significantly frequent in lung cancer patients and results in lung
tumorigenesis (24). Similarly, we
also found that GADD45G was downregulated by PPARG in lung AC.
Moreover, PPARG is a transcriptional factor which mediates
pleiotropic effects, including the regulation of genes involved in
lipid metabolism, such as LPL, which is a component of the
PPAR signaling pathway. PPARG and the 9-cis retinoic acid receptor
(RXR) heterodimers bind to the promoter sequence (−169
TGCCCTTTCCCCC −157) of the LPL gene and thus promote the
transcriptional activation of the LPL gene (25,26).
Higher LPL levels accelerate the growth of cancer cells (27) and predict shorter NSCLC patient
survival times (28).
ETV4 (also known as E1AF), which binds to the
enhancer elements of the adenovirus type 5 E1A gene, is a TF of the
ets oncogene family (29). ETV4 is
expressed in NSCLC cells. Significantly, ETV4-transfected NSCLC
cells show a 2-fold increase in cell motility and invasion compared
with parental and vector-transfected control cells (30). ETV4 is able to upregulate multiple
MMP genes that contribute to the malignant phenotype of
cancer cells by inducing invasive and metastatic activities
(31). A previous study has shown
that the ERK-ETV4-MMP1 axis is upregulated in esophageal AC cells
and is a potentially significant driver of the metastatic
progression of esophageal ACs (32). In the current study, our results
revealed that MMP9 expression was upregulated by
ETV4, which may be involved in the development and invasion
of lung AC.
FLI1 is a TF of the ETS family, defined by a highly
conserved DNA-binding domain (33). Its clinical role is most evident in
human Ewing’s sarcoma in which it is fused with EWS. It has been
shown that transduction of the gene EWSR1-FLI1 transforms
NIH3T3 cells and that mutants containing a deletion in either the
EWS domain or the DNA-binding domain in FLI1 lose this ability
(34,35). Significantly, EWS-FLI1 binds to the
second positive regulatory element of the TGF-b RII promoter, a
putative tumor suppressor gene in the TGF-β signaling pathway, and
suppresses transcription of the TGF-b RII gene at the mRNA
and protein levels (36).
Antisense to EWSR1-FLI1 in ES cell lines positive for this gene
fusion restores TGF-β RII expression (37) which blocks tumorigenicity. However,
the expression of FLI1 in lung cancer cells is not well
characterized. FLI1 expression is usually detected at lower levels
in certain non-hematopoietic tissues, including the lung. Weak
nuclear immunoreactivity is observed in lung AC (38). Therefore, TGF-β RII expression may
be upregulated to block tumorigenicity. In addition, levels of
CCND3 and FLI1 are also positively correlated, since FLI1 maintains
high levels of CCND3 in erythroblasts, thereby promoting
proliferation over differentiation (39). Our results also suggest that FLI1
upregulates CCND3 expression. Therefore, we also suggest that CCND3
expression is associated with the proliferation of lung AC
cells.
NFκB1 is a subunit of the TF NFκB which is derived
by proteolytic cleavage from the N-terminus of a 105-kDa precursor
protein (40). Activated NFκB
stimulates the expression of genes involved in a wide variety of
biological functions; for example, NFκB target genes, including
chemokine (C-C motif) ligand 19 (CCL19), CCL21,
chemokine (C-X-C motif) ligand 12 (CXCL12), CXCL13
and B-cell-activating factor of the tumour-necrosis-factor
family (BAFF), were markedly upregulated in pancreatic
ductal AC cell lines (41). In
addition, the −300 region of the IL-1B promoter contains a
functional NFKB binding site composed of the decamer sequence
5′-GGGAAAATCC-3′ (42). Therefore,
activated NFκB may also induce the expression of IL-1B, which is an
important cytokine involved in inflammatory and immune diseases,
including various cancers (43).
CEBPB is a bZIP TF which may bind as a homodimer to
certain DNA regulatory regions or form heterodimers with the
related proteins CEBP-α, CEBP-δ, and CEBP-γ. CEBPB protein is
important in the regulation of genes involved in immune and
inflammatory responses. It has been shown to activate the IL-6
promoter and induce elevated IL-6 expression levels (44), which are frequently observed in
human lung ACs (45). A further
study has revealed that the regulation of the IL-6 promoter by
CEBPB is completely dependent upon co-operative functions with NFκB
in autocrine human prostate cancer cells (46), suggesting a model in which the bZIP
protein primarily functions to augment the activity of NF-κB.
The TAL1 (or SCL) gene encodes a basic
helix-loop-helix (bHLH) TF that has been demonstrated to be
significant in hematopoiesis and vasculogenesis (47). Previous studies have revealed that
the expression of the SCL interrupting locus gene is increased in
lung ACs and promotes metastatic spread, which may result from the
controlling effect on its downstream gene, SCL (48). KDR (VERFR2) is one of the two VEGF
receptors which is critical for mediating angiogenic endothelial
cell responses via the VEGF pathway (49). Higher levels of soluble VEGFR2 have
been observed in NSCLC patients compared with healthy controls
(50). E-box protein E2-2 blocks
endothelial cell activation via perturbation of VEGFR2 promoter
activity (51). However, TAL1/SCL
relieves the E2-2-mediated repression of VEGFR2 reporter activity
in endothelial cells by interacting with certain DNA sequences of
E2-2 (52).
A basic understanding of the mechanisms underlying
AC is valuable. A deeper understanding of TFs and their regulated
genes remains an area of intense study. Our present findings shed
new light on the complex interacting mechanisms of TFs and their
regulated genes in lung AC. These results may provide potential
therapeutic targets for lung AC treatment.
References
|
1
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality, and prevalence across five
continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar
|
|
2
|
Travis WD, Travis LB and Devesa SS: Lung
cancer. Cancer. 75(Suppl 1): 191–202. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Riely GJ, Kris MG, Rosenbaum D, et al:
Frequency and distinctive spectrum of KRAS mutations in never
smokers with lung adenocarcinoma. Clin Cancer Res. 14:5731–5734.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marks JL, Broderick S, Zhou Q, et al:
Prognostic and therapeutic implications of EGFR and KRAS mutations
in resected lung adenocarcinoma. J Thorac Oncol. 3:111–116. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kosaka T, Yatabe Y, Onozato R, Kuwano H
and Mitsudomi T: Prognostic implication of EGFR, KRAS, and TP53
gene mutations in a large cohort of Japanese patients with
surgically treated lung adenocarcinoma. J Thorac Oncol. 4:22–29.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gill RK, Yang SH, Meerzaman D, et al:
Frequent homozygous deletion of the LKB1/STK11 gene in non-small
cell lung cancer. Oncogene. 30:3784–3791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ding L, Getz G, Wheeler DA, et al: Somatic
mutations affect key pathways in lung adenocarcinoma. Nature.
455:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao SP, Mark KG, Leslie K, et al:
Mutations in the EGFR kinase domain mediate STAT3 activation via
IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hiramatsu M, Ninomiya H, Inamura K, et al:
Activation status of receptor tyrosine kinase downstream pathways
in primary lung adenocarcinoma with reference of KRAS and EGFR
mutations. Lung Cancer. 70:94–102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stearman RS, Dwyer-Nield L, Zerbe L, et
al: Analysis of orthologous gene expression between human pulmonary
adenocarcinoma and a carcinogen-induced murine model. Am J Pathol.
167:1763–1775. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wachi S, Yoneda K and Wu R:
Interactome-transcriptome analysis reveals the high centrality of
genes differentially expressed in lung cancer tissues.
Bioinformatics. 21:4205–4208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smyth GK: Linear models and empirical
bayes methods for assessing differential expression in microarray
experiments. Stat Appl Genet Mol Biol. 3:Article 32004.
|
|
13
|
Irizarry RA, Hobbs B, Collin F, et al:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar
|
|
14
|
Wingender E: The TRANSFAC project as an
example of framework technology that supports the analysis of
genomic regulation. Brief Bioinform. 9:326–332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang C, Xuan Z, Zhao F and Zhang MQ:
TRED: a transcriptional regulatory element database, new entries
and other development. Nucleic Acids Res. 35:D137–D140. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kanehisa M: The KEGG database (Review).
Novartis Found Symp. 247:91–101. 2002. View Article : Google Scholar
|
|
17
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
18
|
Chang TH and Szabo E: Induction of
differentiation and apoptosis by ligands of peroxisome
proliferator-activated receptor γ in non-small cell lung cancer.
Cancer Res. 60:1129–1138. 2000.
|
|
19
|
Theocharis S, Kanelli H, Politi E, et al:
Expression of peroxisome proliferator activated receptor-gamma in
non-small cell lung carcinoma: correlation with histological type
and grade. Lung Cancer. 36:249–255. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Keshamouni VG, Reddy RC, Arenberg DA, et
al: Peroxisome proliferator-activated receptor-γ activation
inhibits tumor progression in non-small-cell lung cancer. Oncogene.
23:100–108. 2004.
|
|
21
|
Hall PA, Kearsey JM, Coates PJ, Norman DG,
Warbrick E and Cox LS: Characterisation of the interaction between
PCNA and Gadd45. Oncogene. 10:2427–2433. 1995.PubMed/NCBI
|
|
22
|
Zhan Q, Antinore MJ, Wang XW, et al:
Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase
activity by the p53-regulated protein Gadd45. Oncogene.
18:2892–2900. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bruemmer D, Yin F, Liu J, et al:
Regulation of the growth arrest and DNA damage-inducible gene 45
(GADD45) by peroxisome proliferator-activated receptor γ in
vascular smooth muscle cells. Circ Res. 93:e38–e47. 2003.
|
|
24
|
Na YK, Lee SM, Hong HS, Kim JB, Park JY
and Kim DS: Hypermethylation of growth arrest DNA-damage-inducible
gene 45 in non-small cell lung cancer and its relationship with
clinicopathologic features. Mol Cells. 30:89–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schoonjans K, Peinado-Onsurbe J, Lefebvre
AM, et al: PPARalpha and PPARgamma activators direct a distinct
tissue-specific transcriptional response via a PPRE in the
lipoprotein lipase gene. EMBO J. 15:5336–5348. 1996.PubMed/NCBI
|
|
26
|
Li L, Beauchamp MC and Renier G:
Peroxisome proliferator-activated receptor alpha and gamma agonists
upregulate human macrophage lipoprotein lipase expression.
Atherosclerosis. 165:101–110. 2002. View Article : Google Scholar
|
|
27
|
Kuemmerle NB, Rysman E, Lombardo PS, et
al: Lipoprotein lipase links dietary fat to solid tumor cell
proliferation. Mol Cancer Ther. 10:427–436. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cerne D, Zitnik IP and Sok M: Increased
fatty acid synthase activity in non-small cell lung cancer tissue
is a weaker predictor of shorter patient survival than increased
lipoprotein lipase activity. Arch Med Res. 41:405–409. 2010.
View Article : Google Scholar
|
|
29
|
Shindoh M, Higashino F and Kohgo T: E1AF,
an ets-oncogene family transcription factor (Review). Cancer Lett.
216:1–8. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hiroumi H, Dosaka-Akita H, Yoshida K, et
al: Expression of E1AF/PEA3, an Ets-related transcription factor in
human non-small-cell lung cancers: Its relevance in cell motility
and invasion. Int J Cancer. 93:786–791. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Higashino F, Yoshida K, Noumi T, Seiki M
and Fujinaga K: Ets-related protein E1A-F can activate three
different matrix metalloproteinase gene promoters. Oncogene.
10:1461–1463. 1995.PubMed/NCBI
|
|
32
|
Keld R, Guo B, Downey P, Gulmann C, Ang YS
and Sharrocks AD: The ERK MAP kinase-PEA3/ETV4-MMP-1 axis is
operative in oesophageal adenocarcinoma. Mol Cancer. 9:3132010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Truong A and Ben-David Y: The role of
Fli-1 in normal cell function and malignant transformation
(Review). Oncogene. 19:6482–6489. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
May WA, Arvand A, Thompson AD, Braun BS,
Wright M and Denny CT: EWS/FLI1-induced manic fringe renders NIH
3T3 cells tumorigenic. Nat Genet. 17:495–497. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thompson AD, Teitell MA, Arvand A and
Denny CT: Divergent Ewing’s sarcoma EWS/ETS fusions confer a common
tumorigenic phenotype on NIH3T3 cells. Oncogene. 18:5506–5513.
1999.
|
|
36
|
Im YH, Kim HT, Lee C, et al: EWS-FLI1,
EWS-ERG, and EWS-ETV1 oncoproteins of Ewing tumor family all
suppress transcription of transforming growth factor β type II
receptor gene. Cancer Res. 60:1536–1540. 2000.PubMed/NCBI
|
|
37
|
Hahm KB, Cho K, Lee C, et al: Repression
of the gene encoding the TGF-β type II receptor is a major target
of the EWS-FLI1 oncoprotein. Nat Genet. 23:222–227. 1999.
|
|
38
|
Rossi S, Orvieto E, Furlanetto A, Laurino
L, Ninfo V and Dei Tos AP: Utility of the immunohistochemical
detection of FLI-1 expression in round cell and vascular neoplasm
using a monoclonal antibody. Mod Pathol. 17:547–552. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pereira R, Quang CT, Lesault I, Dolznig H,
Beug H and Ghysdael J: FLI-1 inhibits differentiation and induces
proliferation of primary erythroblasts. Oncogene. 18:1597–1608.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Grumont RJ, Fecondo J and Gerondakis S:
Alternate RNA splicing of murine nfkb1 generates a nuclear isoform
of the p50 precursor NF-kappa B1 that can function as a
transactivator of NF-kappa B-regulated transcription. Mol Cell
Biol. 14:8460–8470. 1994.PubMed/NCBI
|
|
41
|
Wharry CE, Haines KM, Carroll RG and May
MJ: Constitutive non-canonical NFkappaB signaling in pancreatic
cancer cells. Cancer Biol Ther. 8:1567–1576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hiscott J, Marois J, Garoufalis J, et al:
Characterization of a functional NF-kappa B site in the human
interleukin 1 beta promoter: evidence for a positive autoregulatory
loop. Mol Cell Biol. 13:6231–6240. 1993.PubMed/NCBI
|
|
43
|
Zienolddiny S, Ryberg D, Maggini V, Skaug
V, Canzian F and Haugen A: Polymorphisms of the interleukin-1 β
gene are associated with increased risk of non-small cell lung
cancer. Int J Cancer. 109:353–356. 2004.
|
|
44
|
Hu HM, Tian Q, Baer M, et al: The C/EBP
bZIP domain can mediate lipopolysaccharide induction of the
proinflammatory cytokines interleukin-6 and monocyte
chemoattractant protein-1. J Biol Chem. 275:16373–16381. 2000.
View Article : Google Scholar
|
|
45
|
Yamaji H, Iizasa T, Koh E, et al:
Correlation between interleukin 6 production and tumor
proliferation in non-small cell lung cancer. Cancer Immunol
Immunother. 53:786–792. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xiao W, Hodge DR, Wang L, Yang X, Zhang X
and Farrar WL: Co-operative functions between nuclear factors
NFkappaB and CCAT/enhancer-binding protein-beta (C/EBP-beta)
regulate the IL-6 promoter in autocrine human prostate cancer
cells. Prostate. 61:354–370. 2004. View Article : Google Scholar
|
|
47
|
Tang T, Shi Y, Opalenik SR, et al:
Expression of the TAL1/SCL transcription factor in physiological
and pathological vascular processes. J Pathol. 210:121–129. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Erez A, Perelman M, Hewitt SM, et al: Sil
overexpression in lung cancer characterizes tumors with increased
mitotic activity. Oncogene. 23:5371–5377. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Glubb DM, Cerri E, Giese A, et al: Novel
functional germline variants in the vascular endothelial growth
factor receptor 2 gene (KDR) and their effect on gene expression
and micro-vessel density in lung cancer. Clin Cancer Res.
17:5257–5267. 2011. View Article : Google Scholar
|
|
50
|
Sanmartin E, Jantus Lewintre E, Sirera R,
et al: Soluble vascular endothelial growth factor receptor 2
(VEGFR2): New biomarker in advanced non-small cell lung cancer
(NSCLC)? J Clin Oncol (Suppl). 27:e221082009.
|
|
51
|
Tanaka A, Itoh F, Nishiyama K, et al:
Inhibition of endothelial cell activation by bHLH protein E2–2 and
its impairment of angiogenesis. Blood. 115:4138–4147. 2010.
|
|
52
|
Tanaka A, Itoh F, Itoh S and Kato M:
TAL1/SCL relieves the E2-2-mediated repression of VEGFR2 promoter
activity. J Biochem. 145:129–135. 2009. View Article : Google Scholar : PubMed/NCBI
|