Introduction
Aspirin-exacerbated respiratory disease (AERD) is a
chronic bronchial response to oral intake of non-steroidal
anti-inflammatory drugs (NSAIDs), including aspirin (1). The symptoms of AERD include aspirin
sensitivity, bronchial asthma and chronic rhinosinusitis with nasal
polyposis (2–4). Approximately 20% of adult asthmatics
are known to have aspirin intolerance (5). Thus, AERD is considered as a major
health issue in the epidemiology of asthma.
A hypothesis for AERD pathogenesis states that the
cyclooxygenase (COX)-1 enzyme is inhibited by NSAIDs, including
aspirin, resulting in a decrease in the level of prostaglandin
(PG)-E2 (5). Furthermore, the
decreased amount of PG-E2 induces histamine release from mast cells
that may influence aspirin sensitivity and induce the production of
leukotrienes in the 5-lipoxygenase pathway (5).
CD55, also known as decay accelerating factor (DAF),
is a membrane protein that is associated with the complement
system. In the innate and adaptive immunity, the complement system
plays a key role as a biochemical cascade, clearing pathogens from
the host. However, the complement system is also able to cause
critical damage to the host cell and this leads to immune diseases
affecting the host, including asthma. The complement system is
tightly regulated by several complement control proteins including
the membrane cofactor protein (MCP), C4b-binding protein (C4BP),
factor H (fH) and CD55. Among the complement control proteins, CD55
plays a key role in the regulation of the complement system by
preventing the assembly of the C3bBb complex or accelerating the
disassembly of pre-formed convertase, resulting in a block of the
complement system cascade. This regulation inhibits and limits
production of anaphylatoxins, including C3a, C4a and C5a (6,7).
Although not in asthma, it has been demonstrated that NSAID blocks
CD55 expression and is correlated with the level of COX-1
(8). In line with these studies,
we established a hypothesis that the polymorphisms in CD55 may
cause AERD. Therefore, we examined the genetic association of
CD55 polymorphisms with the risk of AERD.
Materials and methods
Study subjects
A total of 163 subjects with AERD and 429
aspirin-tolerant asthma (ATA) subjects were recruited from the
Asthma Genome Research Center comprising hospitals of
Soonchunhyang, Chonnam, Chungbuk, Seoul national and Chung-Ang
Universities in Korea. Oral aspirin challenge (OAC) was performed
with increasing doses of aspirin (9,10).
Briefly, patients with a history of aspirin hypersensitivity were
provided with 30 mg orally. Respiratory and nasal symptoms, blood
pressure, external signs (urticaria and angioedema) and forced
expiratory volume in 1 sec (FEV1) were documented every
30 min for a period of 1.5 h. In the absence of any indication of
an adverse reaction after 1.5 h, increasing dosages of aspirin (60,
100, 300 and 400 mg) were administered until the patient developed
the reaction and the same measurements were repeated every hour.
Those with no history of aspirin hypersensitivity were started on
100 mg of aspirin and gradually the dosage was increased to 200 mg,
350 mg and 450 mg until the patient developed the reaction. If no
reaction occurred 4 h after the final dose, the test was deemed
negative. Aspirin-induced bronchospasm, reflected by a decline (%)
in FEV1, was calculated as the pre-challenge
FEV1 minus the post-challenge FEV1, divided
by the pre-challenge FEV1. OAC reactions were
categorized into three groups as follows: i) 15% or greater
decrease in FEV1 or nasal reactions (AERD), ii) less
than 15% decrease in FEV1 without naso-ocular or
cutaneous reactions (ATA), or iii) less than 15% decrease in
FEV1 with cutaneous reactions [aspirin-induced urticaria
(AIU)]. All individuals provided informed consent to participate in
the study. The methods were approved by the local ethics committees
of hospital.
Single nucleotide polymorphism (SNP)
selection and genotyping
We selected candidate polymorphic SNPs in the
National Center for Biotechnology Information (NCBI; build 36) and
the International HapMap Project (http://hapmap.ncbi.nlm.nih.gov/) based on the
frequencies in the Asian population and linkage disequilibrium (LD)
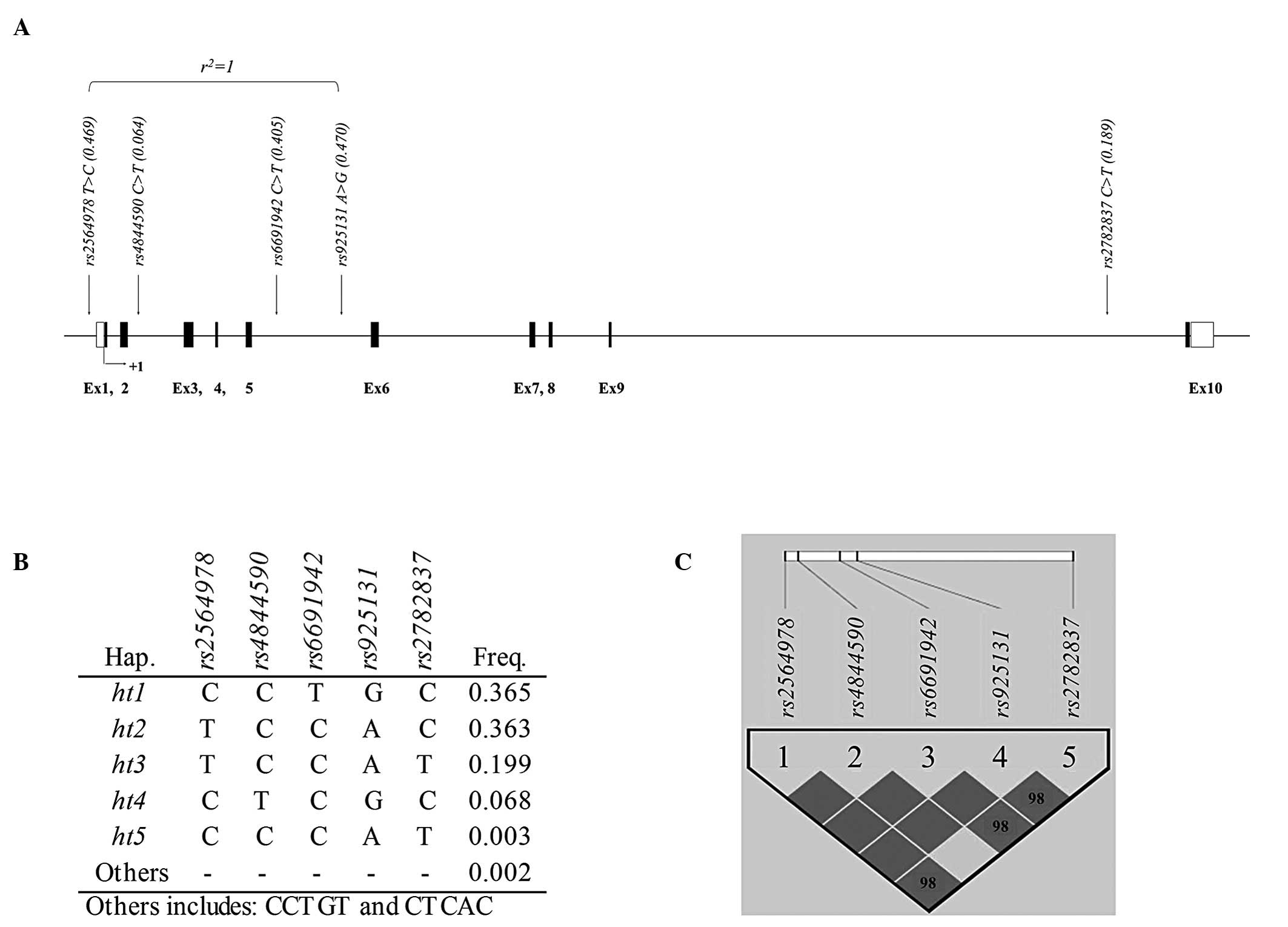
status. For examination of AERD risk association, a total of five
polymorphisms were selected for this study. The location of the
variants is indicated in the genetic map of CD55 as shown in
Fig. 1A. Among the five SNPs, only
one SNP (rs2564978) is located in the promoter region while
the other four SNPs (rs4844590, rs6691942,
rs925131 and rs2782837) are located in introns of
CD55 (Table II). The minor
allele frequencies (MAFs) of all SNPs and allelic variations are
listed in Table II. Genotyping
was carried out with 20 ng of genomic DNA using the TaqMan assay in
the ABI prism 7900HT sequence detection system (Applied Biosystems,
CA, USA) in 163 AERD cases and 429 ATA controls with the assessment
of data quality by duplicate DNAs (n=10).
 | Table IIAssociation analysis of CD55
polymorphisms and haplotypes with risk of AERD. |
Table II
Association analysis of CD55
polymorphisms and haplotypes with risk of AERD.
| Co-dominant | Dominant | Recessive |
|---|
|
|
|
|
|---|
| Polymorphism | OR (95% CI) | P-value | OR (95% CI) | P-value | OR (95% CI) | P-value |
|---|
| rs2564978
T>C | 1.10
(0.85–1.42) | 0.47 | 0.98
(0.66–1.47) | 0.93 | 1.34
(0.87–2.06) | 0.18 |
| rs4844590
C>T | 1.21
(0.72–2.02) | 0.48 | 1.30
(0.76–2.23) | 0.34 | - | 0.99 |
| rs6691942
C>T | 1.05
(0.81–1.37) | 0.7 | 0.97
(0.66–1.42) | 0.87 | 1.26
(0.78–2.04) | 0.35 |
| rs925131
A>G | 1.09
(0.85–1.42) | 0.5 | 0.97
(0.65–1.46) | 0.9 | 1.33
(0.87–2.05) | 0.19 |
| rs2782837
C>T | 1.14
(0.82–1.56) | 0.44 | 1.09
(0.74–1.61) | 0.66 | 1.61
(0.69–3.75) | 0.27 |
|
CD55_ht1 | 1.06
(0.82–1.38) | 0.65 | 0.98
(0.67–1.43) | 0.91 | 1.28
(0.79–2.08) | 0.31 |
|
CD55_ht2 | 0.81
(0.61–1.07) | 0.14 | 0.85
(0.59–1.23) | 0.39 | 0.57
(0.30–1.08) | 0.08 |
|
CD55_ht3 | 1.14
(0.83–1.57) | 0.41 | 1.11
(0.75–1.64) | 0.61 | 1.60
(0.69–3.72) | 0.27 |
|
CD55_ht4 | 1.21
(0.72–2.02) | 0.48 | 1.30
(0.76–2.23) | 0.34 | - | 0.99 |
Statistical analysis
We calculated LD in all the pairs of biallelic loci
using Lewontin’s D’ (|D’|) (11)
and r2. PHASE algorithm (ver. 2.0), developed by as
previously described by Stephens et al, was used for
inferring haplotypes (12).
Associations of genotypes and haplotypes in the CD55 gene
with AERD were calculated using logistic analysis adjusted for age,
gender, smoking status, atopy and body mass index (BMI) as
covariates. We also performed linear regression analysis to
determine the differences in the rates of decline in
FEV1 following aspirin challenge among the genotypes and
haplotypes. The data were adjusted, managed and analyzed using
Statistical Analysis System (SAS) version 9.1 (SAS Inc., Cary, NC,
USA). Statistical power was calculated by PGA (Power for Genetic
Association analysis) software with 5.4% disease prevalence,
relative risk of 1.3 and MAFs of our subjects and subject sizes
(13,14).
Results
Patients
A total of 592 asthma patients including 163 AERD
cases and 429 ATA controls were recruited for analysis. The
characteristics of the patients are summarized in Table I. Among the factors of diagnosis,
percentage of FEV1 fall rate by aspirin provocation
showed significant differences between the AERD and ATA groups
(24.63±16.11 and 3.54±4.85, respectively, P<0.0001; Table I). In addition, predicted
FEV1 and amount of PC20 methacholine also proved to be
significantly different between AERD and ATA patients (P=0.05). The
average value of predicted FEV1 was 87.58±16.94 in AERD
patients and 91.66±16.87 in ATA patients (Table I), while the amount of PC20
methacholine was 5.02±7.83 and 6.91±8.90 in AERD and ATA patients
(Table I). For an adequate
logistic regression analysis, gender, percentage of current smoking
status, existence of atopy and BMI were adjusted.
| Table IClinical profiles for association
analysis between aspirin-exacerbated respiratory disease and
control subjects. |
Table I
Clinical profiles for association
analysis between aspirin-exacerbated respiratory disease and
control subjects.
| Clinical profile | Total no. of
subjects | AERD | ATA |
|---|
| Number of
subjects | 592 | 163 | 429 |
| Mean age
(range)a,c | 46.15
(15.40–77.88) | 43.13
(17.22–72.73) | 47.30
(15.40–77.88) |
| Height (cm) | 160.78±8.63 | 161.72
(143.00–196.00) | 160.42
(140.00–199.00) |
| Weight (kg) | 62.81±10.84 | 61.25±10.38 | 63.40±10.97 |
| FEV1
decrease following aspirin challenge (%)b | 9.27±13.24 | 24.63±16.11 | 3.54±4.85 |
| Blood eosinophil
(%) | 6.01±5.73 | 5.96±5.21 | 6.03±5.92 |
| FVC %, predicted | 88.54±14.08 | 90.35±14.04 | 87.85±14.05 |
| FEV1 %,
predicteda | 90.54±16.97 | 87.58±16.94 | 91.66±16.87 |
| PC20, methacholine
(mg/ml)a | 6.43±8.67 | 5.02±7.83 | 6.91±8.90 |
| Total IgE
(IU/ml) | 357.65±604.09 | 348.60±596.44 | 361.00±607.56 |
| Gender
(male/female) | 206/386 | 59/104 | 147/282 |
| Current Smoker
(%) | 27.70 | 21.47 | 30.07 |
| Positive rate of
Nasal polyP (%)b | 33.83 | 57.89 | 26.06 |
| Positive rate of
skin test (%) | 56.42 | 52.76 | 57.81 |
| Positive rate of
specific IgE (Df, %) | 36.38 | 38.30 | 35.75 |
| Positive rate of
specific IgE (Dp, %) | 44.56 | 45.77 | 44.16 |
Genotype distribution
We also calculated the genotype distributions and
all loci were in Hardy-Weinberg equilibrium (Table IV, P>0.05). In addition, we
established an LD plot and haplotypes using the five genotyped
variants. LDs between each variant are shown in Fig. 1C and r2=1 is also
displayed in Fig. 1A. Results of
logistic analyses revealed that CD55 variants are not
associated with the risk of AERD. P-values did not reach
statistical significance in all genetic models (P>0.05). These
data with odds ratios are summarized in Table III. In further regression
analysis, we investigated the differences between allele
distributions and the decline of FEV1 by aspirin
provocation. However, we failed to find significant differences
between allele distribution, including haplotypes and risk of AERD.
P-values of each polymorphism showed no association signal higher
than 0.05 in co-dominant, dominant and recessive models. The
results of regression analysis are summarized in Table III.
| Table IVInformation of CD55
polymorphisms, minor allele frequencies of Korean AERD/ATA patients
and controls from other populations. |
Table IV
Information of CD55
polymorphisms, minor allele frequencies of Korean AERD/ATA patients
and controls from other populations.
| Polymorphism | Heterozygosity | HWE | Statistica
power | MAFs of present
study | MAFs of other
ethnic groups |
|---|
|
|
|---|
| AERD | ATA | Caucasian | Chinese | Japanese | African |
|---|
| rs2564978
T>C | 0.498 | 0.387 | 94.52 | 0.485 | 0.459 | 0.228 | 0.558 | 0.413 | 0.013 |
| rs4844590
C>T | 0.120 | 0.767 | 38.41 | 0.074 | 0.059 | 0.305 | 0.044 | 0.089 | 0.167 |
| rs6691942
C>T | 0.482 | 0.552 | 94.36 | 0.411 | 0.400 | 0.398 | 0.395 | 0.488 | 0.190 |
| rs925131
A>G | 0.498 | 0.384 | 94.52 | 0.485 | 0.460 | 0.712 | 0.442 | 0.587 | 0.987 |
| rs2782837
C>T | 0.307 | 0.138 | 81.99 | 0.204 | 0.181 | - | 0.256 | 0.170 | 0.000 |
|
CD55_ht1 | 0.481 | 0.601 | 94.34 | 0.411 | 0.397 | - | - | - | - |
|
CD55_ht2 | 0.451 | 0.837 | 93.35 | 0.313 | 0.361 | - | - | - | - |
|
CD55_ht3 | 0.305 | 0.119 | 81.83 | 0.202 | 0.179 | - | - | - | - |
|
CD55_ht4 | 0.120 | 0.767 | 38.41 | 0.074 | 0.059 | - | - | - | - |
| Table IIIRegression analysis of CD55
polymorphisms and haplotypes with fall rate of FEV1 by
aspirin provocation. |
Table III
Regression analysis of CD55
polymorphisms and haplotypes with fall rate of FEV1 by
aspirin provocation.
| Polymorphism | C/C | C/R | R/R | Pa | Pb | Pc |
|---|
| rs2564978
T>C | 172
(9.30±12.79) | 285
(8.94±13.49) | 135
(9.78±13.26) | 0.88 | 0.87 | 0.66 |
| rs4844590
C>T | 518
(9.17±13.17) | 71
(10.17±13.63) | 3 (−2.07±7.59) | 0.98 | 0.79 | 0.17 |
| rs6691942
C>T | 213
(9.49±13.36) | 279
(8.63±12.85) | 99
(10.41±14.02) | 0.86 | 0.68 | 0.39 |
| rs925131
A>G | 171
(9.32±12.82) | 284
(8.95±13.51) | 135
(9.78±13.26) | 0.88 | 0.87 | 0.67 |
| rs2782837
C>T | 393
(9.08±13.58) | 172
(8.83±11.82) | 25
(13.95±16.30) | 0.40 | 0.76 | 0.10 |
|
CD55_ht1 | 214
(9.48±13.33) | 280
(8.60±12.84) | 98
(10.50±14.07) | 0.83 | 0.69 | 0.36 |
|
CD55_ht2 | 255
(9.51±13.20) | 266
(9.60±13.47) | 71
(6.88±12.25) | 0.35 | 0.70 | 0.18 |
|
CD55_ht3 | 396
(9.09±13.55) | 171
(8.88±11.84) | 25
(13.95±16.30) | 0.36 | 0.70 | 0.10 |
|
CD55_ht4 | 518
(9.17±13.17) | 71
(10.17±13.63) | 3 (−2.07±7.59) | 0.98 | 0.79 | 0.17 |
Discussion
According to a hypothesis that AERD is not
correlated with IgE, immune responses including eosinophil
infiltration are considered as important factors of AERD
pathogenesis (15). Additionally,
a previous study demonstrated that leukotriene molecules are
correlated with an expression of complement receptors, including
C3b (16). C3a and C5a play key
roles in attraction of immune cells, including eosinophil (17). In addition, another group
demonstrated that the inflammatory cytokines IL-4 and IL-1β enhance
the expression and release of CD55 (18). The cytokine is able to activate
neutrophils, leading to CD55 expression on neutrophils (19). Therefore, the concentration of CD55
protein may affect the activity of eosinophils and neutrophils,
which are important in AERD pathogenesis.
Previous studies have demonstrated that the
pro-inflammatory mediators, including lipopolysaccharide, tumor
necrosis factor-α and IL-1β, regulate the expression level of
CD55(18,20,21).
In addition, it has been reported that the cyclooxygenase (COX)-2
pathway expressed in the inflammatory cells is also upregulated by
the pro-inflammatory mediators (22). PG-E2, which is generated from the
COX-1 and COX-2 pathways, modulates immune function through a
variety of mechanisms (23,24).
Thus, CD55 may affect the risk of AERD through the COX
pathways.
The study by Kim et al(9) shows only the results from a
co-dominant model of logistic analysis. However, in order to
investigate a possible association between the CD55 SNPs and
the risk of AERD in this study, we carried out more comprehensive
analyses using dominant and recessive models. In addition, we
performed a regression analysis using the fall rate of
FEV1 due to aspirin provocation. However, although we
have performed more thorough analyses, all of our results did not
support our hypothesis. However, the comparison between allele
frequencies in our subjects and other ethnic groups from dbSNP
database shows that the allele frequencies of the SNPs were similar
within the Asian population, but different when compared to those
of Caucasian and African subjects (Table IV). Therefore, further studies
with other ethnic groups are required to validate the exact
function of CD55 polymorphisms in AERD.
In the present study, we used only common
polymorphisms which have frequencies higher than 0.05. Thus, in
order to validate the exact function of polymorphisms in
CD55, replication studies using rare variants which have
frequencies lower than 0.05 may be required. Additionally, the
average statistical power of the present study is 79.08%,
indicating that our sample size was not sufficient for this
analysis. However, rarity of AERD makes it difficult to recruit
sufficient patients for an analysis in Korean asthma cohorts. Thus,
further study using larger independent groups and/or meta-analysis
may be required to investigate the functions of CD55 gene
further. In addition, a comparison between asthmatics and a healthy
normal control is also needed for an full understanding of the
gene.
In conclusion, we identified five SNPs in the human
CD55 gene and explored the effect of polymorphisms in
aspirin-exacerbated respiratory disease subjects in a Korean
population. However, statistical analyses showed no association
between polymorphisms in the promoter and intron and fall rate of
FEV1. Despite the importance of CD55 protein in the
immune system, we failed to find convincing evidence of association
between polymorphisms in CD55 and development of AERD. Due
to various limitations of the present study, further studies using
a large independent population and rare alleles may be required.
Although our P-values did not show significance, results from this
study may be useful for future research in human airway
diseases.
We failed to find evidence of association between
polymorphisms in CD55 and risk of AERD in both logistic and
regression analysis. However, various limitations of the present
study, further studies using large independent population and rare
alleles may be required. Although our P-values did not show
significance, results from this study may be useful in the etiology
of AERD and other bronchial diseases.
Acknowledgements
This study was supported by a National Research
Foundation of Korea (NRF) grant funded by the Korean government
(MEST; no. 2009-0080157 and no. 2010-0011206). This study was
supported by a grant of the Korea Healthcare Technology R&D
Project, Ministry for Health, Welfare and Family Affairs, Republic
of Korea (A010249). The DNA samples were generously provided by
Soonchunhyang University, Bucheon Hospital Biobank (Seoul, Korea),
a member of the National Biobank of Korea, supported by the
Ministry of Health, Welfare and Family Affairs, Republic of
Korea.
References
|
1
|
Widal F, Abrami P and Lermoyez J:
Anaphylaxie et idiosyncrasie. 1992 [Anaphylaxis and idiosyncrasy
1992]. Allergy Proc. 14:371–376. 1993. View Article : Google Scholar
|
|
2
|
Samter M and Beers RF Jr: Concerning the
nature of intolerance to aspirin. J Allergy. 40:281–293. 1967.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stevenson DD, Sanchez-Borges M and
Szczeklik A: Classification of allergic and pseudoallergic
reactions to drugs that inhibit cyclooxygenase enzymes. Ann Allergy
Asthma Immunol. 87:177–180. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pawankar R: Nasal polyposis: an update:
editorial review. Curr Opin Allergy Clin Immunol. 3:1–6. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Szczeklik A and Stevenson DD:
Aspirin-induced asthma: advances in pathogenesis, diagnosis, and
management. J Allergy Clin Immunol. 111:913–921; quiz 922. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liszewski MK, Farries TC, Lublin DM,
Rooney IA and Atkinson JP: Control of the complement system. Adv
Immunol. 61:201–283. 1996. View Article : Google Scholar
|
|
7
|
Turnberg D and Botto M: The regulation of
the complement system: insights from genetically-engineered mice.
Mol Immunol. 40:145–153. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Akhtar MS, Kousar F, Masood M, Fatimi S
and Kokab: Evaluation of paclitaxel and carboplatin versus
combination chemotherapy with fluorouracil, doxorubicin and
cyclophosphamide as a neoadjuvant therapy in patients with
inoperable breast cancer. J Coll Physicians Surg Pak. 20:748–752.
2010.
|
|
9
|
Kim BS, Park SM, Uhm TG, et al: Effect of
single nucleotide polymorphisms within the interleukin-4 promoter
on aspirin intolerance in asthmatics and interleukin-4 promoter
activity. Pharmacogenet Genomics. 20:748–758. 2010.PubMed/NCBI
|
|
10
|
Nizankowska-Mogilnicka E, Bochenek G,
Mastalerz L, et al: EAACI/GA2LEN guideline: aspirin provocation
tests for diagnosis of aspirin hypersensitivity. Allergy.
62:1111–1118. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hedrick PW: Gametic disequilibrium
measures: proceed with caution. Genetics. 117:331–341.
1987.PubMed/NCBI
|
|
12
|
Stephens M, Smith NJ and Donnelly P: A new
statistical method for haplotype reconstruction from population
data. Am J Hum Genet. 68:978–989. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Menashe I, Rosenberg PS and Chen BE: PGA:
power calculator for case-control genetic association analyses. BMC
Genet. 9:362008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hedman J, Kaprio J, Poussa T and Nieminen
MM: Prevalence of asthma, aspirin intolerance, nasal polyposis and
chronic obstructive pulmonary disease in a population-based study.
Int J Epidemiol. 28:717–722. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nasser SM, Pfister R, Christie PE, et al:
Inflammatory cell populations in bronchial biopsies from
aspirin-sensitive asthmatic subjects. Am J Respir Crit Care Med.
153:90–96. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nagy L, Lee TH, Goetzl EJ, Pickett WC and
Kay AB: Complement receptor enhancement and chemotaxis of human
neutrophils and eosinophils by leukotrienes and other lipoxygenase
products. Clin Exp Immunol. 47:541–547. 1982.PubMed/NCBI
|
|
17
|
Taube C, Rha YH, Takeda K, et al:
Inhibition of complement activation decreases airway inflammation
and hyperresponsiveness. Am J Respir Crit Care Med. 168:1333–1341.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nasu J, Mizuno M, Uesu T, et al:
Cytokine-stimulated release of decay-accelerating factor (DAF;CD55)
from HT-29 human intestinal epithelial cells. Clin Exp Immunol.
113:379–385. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Berger M and Medof ME: Increased
expression of complement decay-accelerating factor during
activation of human neutrophils. J Clin Invest. 79:214–220. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heine H, El-Samalouti VT, Nötzel C, et al:
CD55/decay accelerating factor is part of the
lipopolysaccharide-induced receptor complex. Eur J Immunol.
33:1399–1408. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Andoh A, Fujiyama Y, Sumiyoshi K, Sakumoto
H, Okabe H and Bamba T: Tumour necrosis factor-alpha up-regulates
decay-accelerating factor gene expression in human intestinal
epithelial cells. Immunology. 90:358–363. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stevenson DD: Aspirin sensitivity and
desensitization for asthma and sinusitis. Curr Allergy Asthma Rep.
9:155–163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Harris SG, Padilla J, Koumas L, Ray D and
Phipps RP: Prostaglandins as modulators of immunity. Trends
Immunol. 23:144–150. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goodwin JS and Ceuppens JL: Effect of
nonsteroidal antiinflammatory drugs on immune function. Semin
Arthritis Rheum. 13:134–143. 1983. View Article : Google Scholar : PubMed/NCBI
|