Introduction
Congenital 3β-hydroxysteroid dehydrogenase (3β-HSD)
deficiency is an autosomal recessive hereditary disease and one of
the causes of congenital adrenal hyperplasia (1,2). In
humans, 3β-HSD has two isozymes that can catalyze the oxidative
conversion of Δ5 into a Δ4 steroid hormone. Type I and II isozymes
have a homology of 93.5% (3,4).
Congenital 3β-HSD deficiency can be classified as classical or
non-classical, according to clinical manifestations. Classic 3β-HSD
deficiency is caused by a mutation in the Type II 3β-HSD gene, and
patients with this type of deficit have different degrees of
salt-wasting, possible pseudohermaphroditism in males, and normal
sexual differentiation or mild masculinization in females (5,6). The
salt-wasting type of 3β-HSD deficiency is usually diagnosed in
newborns, whereas the non-salt-wasting type is diagnosed prior to
adolescence (7,8).
Although clinical reports are available on 3β-HSD
deficiency cases or 3β-HSD deficiency in families in foreign
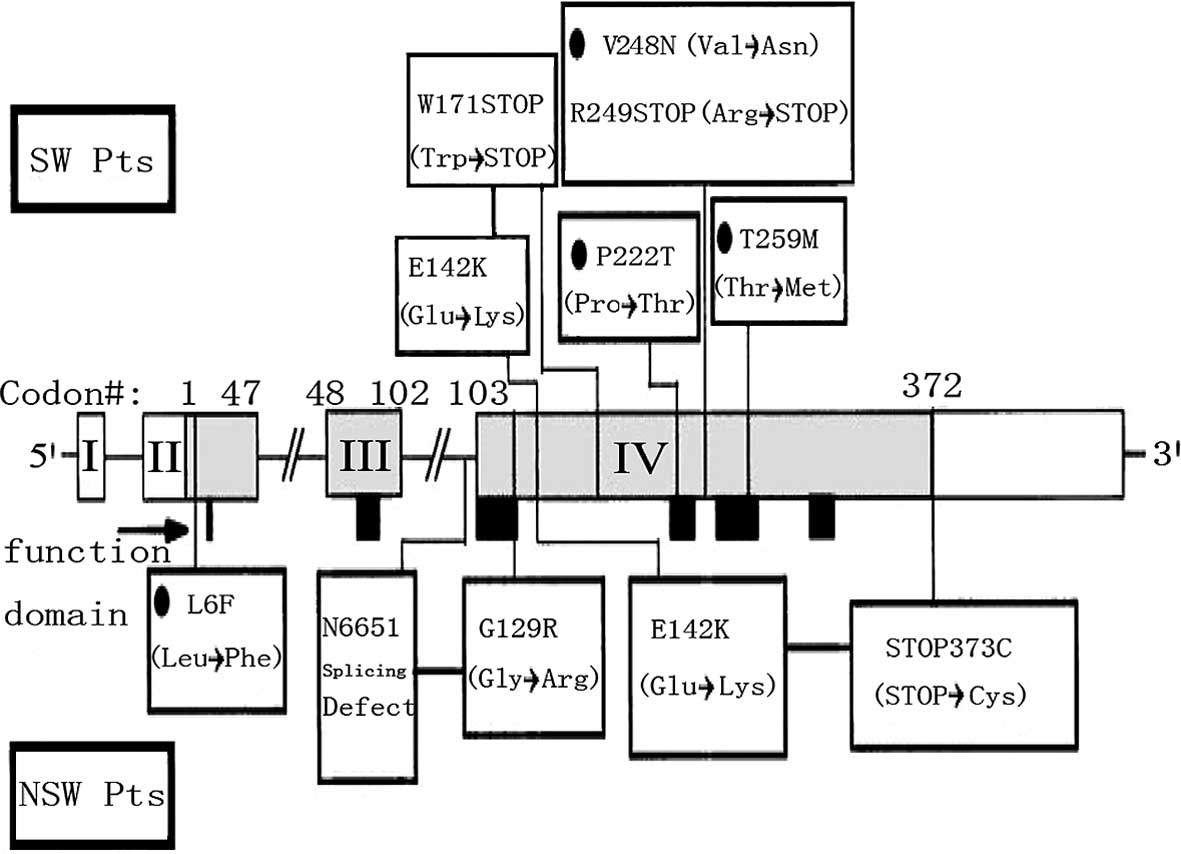
countries (Fig. 1) (9), there is currently no report on the
mutation of the 3β-HSD gene in a Chinese family with adrenocortical
deficiency. In this study, clinical analysis and molecular genetic
research were carried out on a patient suffering from 3β-HSD
deficiency and his family.
Materials and methods
Materials
The patient (male, 17 years old) was hospitalized
because of generalized pigmentation for 17 years, and fever and
convulsions for 4 days. His brown yellow generalized pigmentation
was observed at birth but was not paid attention to because no
abnormalities were found in the patient then in terms of sucking,
breathing or sleeping. At age 3, the patient was found by his
parents to be suffering from shortness of breath after activities,
and he presented to the Southern Anhui Affiliated Hospital. He was
diagnosed with adrenal insufficiency and was administered
prednisone with an initial dose of 15 mg/d. Administration of the
medication was continued at a dose of 5 mg/d for 3 months. Family
members said the disease was somewhat relieved, thus other drugs
were not used after the 3-month period of time. One week prior to
admission, the patient demonstrated the following symptoms: fever,
convulsions and trismus without any oral discharge. However, the
duration was unknown. He was considered to be a possible case of
encephalitis at a local hospital and was given mannitol for
dehydration and appropriate electrolytes. He was also diagnosed
with hepatic and renal dysfunction. No abnornalities were found by
head computed tomography (CT). Blood gas analysis indicated he had
Type I respiratory failure and a mask ventilator was used to
facilitate his breathing. The patient visited our hospital because
his disease was not significantly relieved. Chest scan and
B-ultrasound showed increased markings in bilateral lungs and no
abnormalities in the liver, gallbladder, pancreas, spleen,
bilateral kidneys and adrenal glands. The patient was diagnosed
with adrenal insufficiency and admitted to our hospital. His family
history is as follows: the patient’s paternal grandmother and
maternal grandmother are cousins, his healthy parents’ marriage is
consanguineous, and the patient’s sister also had pigmentation. The
following were the results of the physical examination: height, 173
cm; poor nutrition; weight, 62.5 kg; generalized pigmentation,
especially in the abdomen and waist; obvious oral mucosa; and
gingival pigmentation. This study was conducted in accordance with
the declaration of Helsinki and with the approval of the Ethics
Committee of the Kunshan People’s Hospital Affiliated to the
Jiangsu University. Written informed consent was obtained from the
participant.
Hormone determination
Enzyme immunoassay (Serono Company, Switzerland) was
conducted to assay follicle stimulating hormone (FSH), luteinizing
hormone (LH), estradiol and testosterone. Radioimmunoassay was
conducted to assay cortisol and aldosterone (DiaSorin Corporation,
Stillwater, MN, USA). An immunoradiometric assay was conducted to
assay adrenocortical hormone (DSL Systems Laboratories, Webster,
TX, USA).
PCR amplification
Venous whole blood (5 ml) was collected from the
patient, his parents, sister, 2 aunts and 3 other healthy physical
examinees. EDTA was used for blood anti-coagulation, and an icebox
was used to transport the samples. Genomic DNA was extracted using
the routine phenol-chloroform method and was stored in a
refrigerator (−20°C). Exons 1, 2, 3 and 4 of the 3β-HSD gene of the
proband were analyzed to locate mutation sites, as well as exon 4
of the 3β-HSD gene of the other family members and the healthy
physical examinees. The primers used for PCR are shown in Table I. The primers were synthesized by
the Shanghai Saibaisheng Gene Technology Co., Ltd. (Shanghai,
China). The primers were diluted to 10 pmol/μl and stored at
−20°C.
 | Table IPrimers for PCR amplification. |
Table I
Primers for PCR amplification.
| Primer | Sequence | Primer | Sequence | Fragment | Length (bp) |
|---|
| F1 |
5′-CAGAGCTCTCCAGGGAAAAATTGCA-3′ | R1 |
5′-TTTACAAAAATTCCATGACCCCACA-3′ | exon 1 | 643 |
| F2 |
5′-GCATAAAGCTCCAGTCCTTCCTCCA-3′ | R2 |
5′-TTGCTAGACAAGGTCAACCTCCCCA-3′ | exon1+2 | 521 |
| F3 |
5′-TATCAGAAAACTTCCCAGCCAGATC-3′ | R3 |
5′-TCTGATCCTCATTTAACCAACTTGT-3′ | exon 3 | 279 |
| F4 |
5′-TGGGATATTTCCTGCACTGTCATC-3′ | R4 |
5′-AGGACCTGGGCTTGTGCCCCTGTTG-3′ | exon 4 | 959 |
| F5 |
5′-GGAAGTAGTGAGCTTCCTACTCAGC-3′ | R5 |
5′-ATGGTGATAGTTGGAAATGAAAGGA-3′ | exon 4 | 707 |
The PCR reaction system contained genomic DNA, 150
ng; upstream primer, 0.625 μl; downstream primer, 0.625 μl; 10X
HotStar buffer, 2.5 μl; dNTP, 0.5 μl; and HotStar DNA polymerase,
0.125 μl. Then, ddH2O was added until the total volume
was 25 μl. The PCR reaction conditions were F1/R1, F4/R4, F5/R5:
95°C for 10 min, 94°C for 1 min, 60°C for 1 min, 72°C for 1 min and
35 cycles of 72°C for 10 min; F2/R2, F3/R3: 95°C for 10 min, 94°C
for 45 sec, 62 °C for 45 sec, 72°C for 1 min and 40 cycles of 72°C
for 10 min. PCR products were assayed using 1.5% agarose gel
electrophoresis, and then dyed with ethidium bromide. Bands were
observed under ultraviolet light.
Sequencing
The PCR products were bidirectionally sequenced by
the Shanghai Invitrogen Biotechnology Co., Ltd., and all amplified
fragments were assayed through bi-directional sequencing.
Results
Laboratory index
Routine blood analysis revealed that RBC was
3.67×1012/l, Hb was 101 g/l, N% was 59.4% and platelets
were 82×109/l. Routine urinalysis showed the following
results: occult blood, −; ketones, +; glucose, −and protein, +.
Routine fecalysis was normal. Liver function showed that ALT was 64
U/l, AST was 197 U/l and albumin was 24 g/l. Renal function
revealed serum creatinine to be 203 μmol/l. Endocrine hormone
analysis showed the following results: cortisol 0.86 μg/dl
(6.2–19.4); adrenocorticotropic hormone (ACTH) 132 pg ml (<46
pg/ml); estradiol 180.8 pmol/l (28.0–156 pmol/l); progesterone 0.39
nmol/l (0.7–4.3 nmol/l); LH 7.62 IU/l (1.7–8.6 IU/l); FSH 1.0 IU/l
(1.50–12.4 IU/l); testosterone 2.53 nmol/l (9.9–27.8 nmol/l);
parathyroid hormone 69.9 pg/ml (14–72 pg/ml); urinary
17-hydroxycorticosteroid 101.6 μmol/24 h (8.3–27.7 μmol/24 h) and
urinary 17-ketosteroid 48.7 μmol/24 h (35–87 μmol/24 h). Thyroid
peroxidase antibody was normal. Thyrotropin receptor antibody was
4.1 IU/l (<10 IU/l). Fasting blood glucose was normal. ENA
Euroassay was negative. Serum calcium was 2.01 mmol/l (2.20–2.70
mmol/l). B-ultrasound showed no obvious abnormalities in the liver,
gallbladder, pancreas, spleen, kidneys or adrenal glands. The
thyroid resembled a 2-leaf tiny follicular nodule. The chest CT
showed a diffused infiltration shadow and a consolidation shadow in
both lungs, suggesting the possible presence of an inflammatory and
pulmonary edema or a bilateral pleural effusion.
Genetic testing
Exons 1-3 of the patient were amplified (primers
F1/R1). No gene mutation was found relative to the normal
nucleotide sequence (GenBank No. GI184396). The patient’s exon 4
(primers F5/R5) was amplified, and homozygous mutations were found
at nucleotide 1088 C (C/T) and nucleotide 1132 C (C/G). His sister
also had homozygous mutations at these sites, whereas his father,
mother and Aunt 1 had heterozygous mutations. Aunt 2 had no
mutations at these sites. One of the 3 healthy physical examinees
had heterozygous mutations at the 2 sites; the other examinees had
no genetic mutations.
Discussion
In the case analyzed in this study, the proband
visited our hospital because of adrenal crisis. At birth, the
proband had generalized pigmentation, obvious oral mucosa
pigmentation and gingival pigmentation. He was diagnosed with
adrenal insufficiency at age 3. His sister had similar symptoms.
Their paternal and maternal grandmothers were cousins, thus their
parents’ marriage was consanguineous. Therefore, the patient’s
disease may be both congenital and familial.
Cortisol synthesis requires cholesterol 20- and
22-lyase, CYP21, CYP11B, 3β-HSD and CYP17. Mutation in any of the
genes encoding these enzymes may lead to deficiency in enzyme
activity, causing different degrees of adrenal failure (10). CYP21 deficiency is the most common
type of congenital adrenal hyperplasia, followed by 3β-HSD
deficiency. CYP17 and 3β-HSD deficiencies are able to decrease the
levels of 3 types of adrenal cortex hormones. CYP21 and CYP11B
deficiencies decrease only the synthesis of cortisol and
aldosterone, but increase the synthesis of sex hormones, which can
lead to spurious sexual precocity in males and masculinization.
CYP18 and 18-oxidase deficiencies decrease only the level of
aldosterone and do not affect the levels of cortisol and sex
hormones.
The physiological role of 3β-HSD is to catalyze the
dehydrogenation of 3β-hydroxy steroids, of pregnenolone to generate
progesterone, of 17α-hydroxypregnenolone to generate
17α-hydroxyprogesterone and of dehydroepiandrosterone to generate
Δ4-androstenedione. The 3β-HSD deficiency can lead to the
accumulation of pregnenolone, 17α-hydroxy pregnenolone and
dehydroepiandrosterone (DHEA), and can decrease the synthesis of
cortisol, aldosterone and testosterone (11–16).
Mineralocorticosteroid and glucocorticosteroid deficiencies lead to
clinical symptoms, such as salt loss, chronic adrenal dysfunction
and mild masculinization (17) as
fetal adrenal glands secrete excessive DHEA, and some DHEA can be
translated into the testosterone through extra-adrenal paths. In
some female patients manifesting masculinization, the level of
17-OHP is high due to the active effect of extra-adrenal 3β-HSD. If
enzyme deficiencies affect the adrenal and sex glands, male embryos
are not likely to secrete enough testosterone, which would result
in incomplete masculinization at birth, with hypospadias,
cryptorchidism and even male pseudohermaphroditism (3,18).
In this report, the cortisol level of the proband was 0.86 μg/dl,
which was below the normal range of 6.2–19.4 μg/dl. The patient’s
ACTH level was 132 pg/ml, whereas the normal level is less than 46
pg/ml. The testosterone level of the proband was 2.53 nmol/l, which
was significantly below the normal range of 9.9–27.8 nmol/l. His
progesterone level was 0.39 nmol/l, which was slightly below the
normal range of 0.4–3.1 nmol/l. The levels of cortisol,
testosterone and progesterone in the patient were lower than
normal, thus he was considered to have congenital adrenal
hyperplasia caused by 3β-HSD.
3β-HSD deficiency is an autosomal recessive,
inherited disease caused by 3β-HSD gene mutation (19–22).
In addition to clinical diagnosis, genetic diagnosis is also
important in ascertaining the existence of 3β-HSD deficiency. In
this report, all the amplified products were analyzed by direct
sequencing. Four exons of the 3β-HSD gene and their flanking
sequences were amplified and sequenced. The method employed was
simple and convenient. Although a small number of DNA sequences
showed impure peaks, the results were reliable as bi-directional
sequencing was performed. By comparing the sequencing results of
the family members mentioned in this study with the sequences of
corresponding normal exons of the 3β-HSD gene (GenBank Accession
No. GI184396), we discovered the following: the family members with
abnormal phenotype had a transition mutation (C/T) at nucleotide
1088 and a transversion mutation (C/G) at nucleotide 1132 in exon 4
of the 3β-HSD gene; both mutations were homozygous. The sequencing
of family members with normal phenotype revealed that the father,
mother and Aunt 1 had heterozygous mutations, whereas Aunt 2 did
not have mutations at the 2 sites. The mutation discovered in this
family has not yet been reported in China or abroad.
In this report, the family members with abnormal
phenotype had homozygous mutations at the 2 sites mentioned above.
The parents and Aunt 1 also had heterozygous mutations at the 2
sites. In addition, genomic DNA was extracted from the venous blood
of 3 healthy physical examinees, and exon 4 of their 3β-HSD gene
was also amplified. One of the examinees had heterozygous mutations
at the 2 sites, in contrast to the other 2 examinees. Therefore,
the mutations in the 2 sites may be linked mutations.
Nucleotide 1088 in exon 4 of the 3β-HSD gene had
transitional mutations (C/T) while nucleotide 1132 had
transversional mutations (C/G). The two mutations were located at
the 3′ end of exon 4, an untranslated region, suggesting that the
activity of 3β-HSD was possibly decreased by the regulation of
translation or transcription. The mechanism by which such new
mutations induce a decrease in the activity of 3β-HSD needs to be
further studied.
References
|
1
|
Rheaume E, Simard J, Morel Y, et al:
Congenital adrenal hyperplasia due to point mutations in the type
II 3β-hydroxysteroid dehydrogenase gene. Nature Genet. 1:239–245.
1992.
|
|
2
|
White PC and New MI: Molecular genetics of
congenital adrenal hyperplasia. Baillieres Clin Endocrinol Metab.
2:941–965. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rheaume E, Lachance Y, Zhao HF, et al:
Structure and expression of a new complementary DNA encoding the
almost exclusive 3 beta-hydroxysteroid dehydrogenase/delta 5-delta
4-isomerase in human adrenals and gonads. Mol Endocrinol.
5:1147–1157. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johannsen TH, Mallet D, Dige-Petersen H,
et al: Delayed diagnosis of congenital adrenal hyperplasia with
salt wasting due to type II 3beta-hydroxysteroid dehydrogenase
deficiency. J Clin Endocrinol Metab. 90:2076–2080. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bongiovanni AM, Eberlein WR, Goldman AS
and New M: Disorders of adrenal steroid biogenesis. Recent Prog
Horm Res. 23:375–449. 1967.PubMed/NCBI
|
|
6
|
Simard J, Rheaume E, Mebarki F, et al:
Molecular basis of human 3β-hydroxysteroid dehydrogenase
deficiency. J Steroid Biochem Mol Biol. 53:127–138. 1995.
|
|
7
|
Reiter JC, Leveau P, Tardy V, Forest MG
and Morel Y: Diagnostic criteria for the diagnosis of
3β-hydroxysteroid dehydrogenase (3β-HSD) deficiency: lessons from
hormonal and molecular studies in 2 girls presenting with premature
pubarche. Horm Res. 53:762002.
|
|
8
|
Simard J, Moisan AM and Morel Y:
Congenital adrenal hyperplasia due to 3beta-hydroxysteroid
dehydrogenase/Delta(5)-Delta(4) isomerase deficiency. Semin Reprod
Med. 20:255–276. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lutfallah C, Wang W, Mason JI, et al:
Newly proposed hormonal criteria via genotypic proof for type II
3beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol
Metab. 87:2611–2622. 2002.PubMed/NCBI
|
|
10
|
Ritzen EM, Lajic S and Wedell A: How can
molecular biology contribute to the management of congenital
adrenal hyperplasia? Horm Res. 53:34–37. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luu the V, Lachance Y, Labrie C, et al:
Full length cDNA structure and deduced amino acid sequence of human
3b-hydroxy-5-ene steroid dehydrogenase. Mol Endocrinol.
3:1310–1312. 1989.PubMed/NCBI
|
|
12
|
Cherradi N, Defay G and Chambaz EM: Dual
subcellular localization of the 3β-hydroxysteroid dehydrogenase
isomerase: characterization of the mitochondrial enzyme in the
bovine adrenal cortex. J Steroid Biochem Mol Biol. 46:773–779.
1993.
|
|
13
|
Cherradi N, Defaye G and Chambaz EM:
Characterization of the 3 beta-hydroxysteroid dehydrogenase
activity associated with bovine adrenocortical mitochondria.
Endocrinology. 134:1358–1364. 1994.
|
|
14
|
Sauer LA, Chapman JC and Dauchy RT:
Topology of 3β-hydroxy-5-enesteroid dehydrogenase/D5-D4-isomerase
in adrenal cortex mitochondria and microsomes. Endocrinology.
134:751–759. 1994.
|
|
15
|
Simard J, Durocher F, Mebarki F, et al:
Molecular biology and genetics of the 3 beta-hydroxysteroid
dehydrogenase/delta5-delta4 isomerase gene family. J Endocrinol.
150:S189–S207. 1996.PubMed/NCBI
|
|
16
|
Thomas JL, Evans BW, Blanco G, et al:
Site-directed mutagenesis identifies amino acid residues associated
with the dehydrogenase and isomerase activities of human type I
(placental) 3β-hydroxysteroid dehydrogenase/isomerase. J Steroid
Biochem Mol Biol. 66:327–334. 1998.PubMed/NCBI
|
|
17
|
Moisan AM, Ricketts ML, Tardy V, et al:
New insight into the molecular basis of 3beta-hydroxysteroid
dehydroxygenase deficiency: identification of eight mutation in the
HSD3B2 gene eleven patients from seven new families and comparison
of the functional properties of twenty-five mutant enzymes. J Clin
Endocrinol Metab. 84:4410–4425. 1999.
|
|
18
|
Miller WL and Levine LS: Molecular and
clinical advances in congenital adrenal hyperplasia. J Pediatr.
111:1–17. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Speiser PW, Dupont J, Zhu D, et al:
Disease expression and molecular genotype in congenital adrenal
hyperplasia due to 21-hydroxylase deficiency. J Clin Invest.
90:584–595. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang L, SakkaL-Alkaddour H, Chang YT,
Yang X and Pang S: A new compound heterozygous frameshift mutation
in the type II 3β-hydroxysteroid dehydrogenase (3β-HSD) gene caused
salt-wasting 3β-HSD deficiency congenital adrenal hyperplasia. J
Clin Endocrinol Metab. 81:291–295. 1996.
|
|
21
|
Moran C, Potter HD, Reyna R, Boots LR and
Azziz R: Prevalence of 3 beta-hydroxysteroid
dehydrogenase-deficient nonclassic adrenal hyperplasia in
hyperandrogenic women with adrenal androgen excess. Am J Obstet
Gynecol. 181:596–600. 1999. View Article : Google Scholar
|
|
22
|
Alos N, Mosian AM, Ward L, et al: A novel
A10E homozygous mutation in the HSD3B2 gene causing severe
salt-wasting 3beta-hydroxysteroid dehydrogenase deficiency in 46,
XX and 46, XY French-Canadians: evaluation of gonadal function
after puberty. J Clin Endocrinol Metab. 85:1968–1974. 2000.
|