Introduction
Helicobacter pylori (H. pylori)
infection is associated with divergent clinical outcomes that range
from simple asymptomatic gastritis to more serious conditions,
including peptic ulcer disease and gastric neoplasia (1). A number of previous studies have
focused on the role of bacterial virulence factors in the
pathogenesis of these diseases. Although these factors undoubtedly
contribute to the degree of tissue damage, the 2 key outcomes,
gastric cancer and duodenal ulcer disease, have yet to be
distinguished (2). Therefore,
understanding of host genetic factors that may be relevant to this
process must be developed further.
The key pathophysiological event in H. pylori
infection is initiation of a gastric mucosal inflammatory response,
which is mediated and regulated by a large number of
pro-inflammatory cytokines, particularly interleukin (IL)-1β, IL-6
and IL-8 (3,4). Previous studies have demonstrated
that the host genetic polymorphisms of IL-1β are relevant to H.
pylori-associated gastric cancer. Polymorphisms in the IL-1β
gene that correlate with increased levels of the cytokine have been
identified to increase the risk of hypochlorhydria and gastric
atrophy in response to H. pylori infection, therefore
increasing the risk of gastric cancer itself (5–7). To
this end, IL-1β, as a potent pro-inflammatory cytokine, may be
involved in the host response to H. pylori infection. At
present, the molecular mechanisms associated with the correlation
between the risk of gastric cancer and the polymorphisms of IL-1β
remain unclear.
Disturbance of the balance between proliferation and
apoptosis of gastric epithelial cells is considered to interfere
with the integrity of gastric mucosa and promote the development of
gastric carcinogenesis (8,9). Gastric acid hyposecretion also
correlates with increased risk of gastric cancer (5). Therefore, in the present study, we
investigated the effects of exogenous IL-1β on proliferation and
apoptosis of gastric epithelial cells and acid secretion of
isolated rabbit parietal cells in order to explore the role of
IL-1β in H. pylori-associated diseases and to identify the
mechanisms involved in H. pylori-induced gastric
carcinogenesis.
Materials and methods
Gastric epithelial cell culture
The human gastric cancer cell line, AGS, and the
human gastric epithelial cell line, GES-1, were maintained in
RPMI-1640 medium containing 10% FBS and in MCDB-153 medium
supplemented with 10% FBS, respectively, at 37°C with 5%
CO2 and 95% air in a humidified incubator. The 2 cell
lines were preserved in our laboratory. Cells were serum-starved
for 12 h prior to treatment and then treated with either vehicle or
test reagents for the indicated time.
Parietal cell preparation
Rabbit parietal cells were isolated and enriched
from male New Zealand rabbits (2±0.2 kg) using a modification of
previously described methods (10). Briefly, gastric fundic mucosa was
digested with sequential exposure to type I crude collagenase
(30–40 mg/100 ml; Sigma-Aldrich, St. Louis, MO, USA) and EDTA (1–2
mmol/l). Parietal cells were enriched from the crude suspension by
the standard centrifugal elutriation technique using a Beckman JE6B
elutriation system. For selected experiments, further purification
of parietal cells was performed using continuous density gradient
centrifugation with 50% Percoll (Pharmacia, Piscataway, NJ, USA).
Parietal cells were enriched to >70% homogeneity as determined
by hematoxylin and eosin staining and >95% viability as
determined by trypan blue exclusion.
Harvested cells from the parietal cell-enriched
fractions were collected by brief centrifugation and resuspended in
complete culture medium (Ham’s F12/DMEM, 1:1). Cells were cultured
at 37°C in 5% CO2, 95% air for 12 h prior to treatment
and then treated with either vehicle or IL-1β (PeproTech, Rocky
Hill, NJ, USA) for the indicated time.
H. pylori preparation
Cytotoxin-associated gene A (CagA)-positive and
cytotoxin-producing H. pylori (NCTC 11637) strain was used
in the present study. Bacteria were grown under microaerophilic
conditions on Columbia agar plates (supplemented with 8% sheep
blood) for 72 h, harvested and resuspended in RPMI-1640 medium.
Bacterial concentrations were standardized by optical density
measurement at 600 nm and validated by serial dilution.
OD600 of ~1.0 corresponded to a bacterial concentration
of 1.5×108 cfu/ml.
MTT assay
Cell proliferation was analyzed using MTT assay.
Cells were seeded on a 96-well plate at 1.0×104 and
0.5×104 cells/well for GES-1 and AGS cells, respectively
and incubated with increasing concentrations of IL-1β (0.1, 1.0 and
10 ng/ml) for 24 h in serum-free culture medium. Each sample had 6
replicates. MTT (0.5 mg/ml) was added and the reaction mixture was
incubated for 4 h at 37°C. Following MTT incubation, cells were
lysed in 150 μl of 10% DMSO and the absorbance at 490 nm was
measured using an automatic plate reader (Bio-Rad, Hercules, CA,
USA). Viable cell number was expressed as a percentage of control:
MTT assay (% control) = ODtest/ODcontrol.
Assessment of cell apoptosis
AGS and GES-1 cells were treated with either vehicle
or test reagents for 24 h prior to assessment of apoptosis. Cells
floating in the culture medium were collected by centrifugation and
adherent cells were harvested by incubation with 1% trypsin for 1–2
min at 70–80% confluence. Following washing with ice-cold PBS,
cells were suspended in 70% ethanol and kept at 4°C for 30 min.
Fixation was terminated by washing twice with PBS and the cells
were stained with propidium iodide (100 μg/ml) at 4°C for 30 min.
The cell suspension was filtered through 50-μm nylon mesh and DNA
fluorescence was analyzed by flow cytometry (FCM; Beckman Coulter,
Miami, FL, USA). A minimum of 10,000 events were measured per
sample. Apoptosis was detected by the appearance of a sub-G1
fraction of fragmented nuclei in the analysis. Apoptosis was
expressed as a percentage of the control and caluculated as:
apoptotic cell (%)test/apoptotic cell
(%)control.
Detection of mRNA by RT-PCR
Using a RNA extraction kit according to the
manufacturer’s instructions, total cellular RNA was isolated from
AGS/GES-1 and parietal cells, which were treated with vehicle or
IL-1β (10 ng/ml) for indicated times prior to isolation. RNA (2 μg)
from each sample was reverse transcribed using Supercript II RT
system (Invitrogen Life Technologies, Carlsbad, CA, USA) in a total
reaction volume of 20 μl and the resulting cDNA was amplified by
PCR. The PCR primer sequences (F, forward; R, reverse) and PCR
product size were as follows: cyclooxygenase-2 (COX-2)-F,
5′-TTCAAATGAGATTGTGGGAAAATTGCT-3′ and COX-2-R,
5′-AGATCATCTCTGCCTGAGTATCTT-3′, 305 bp; β-actin-F,
5′-CCAGAGCAAGAGAGGTATCC-3′, β-actin-R, 5′-CTGTGGTGGTGAAGCTGTAG-3′,
463 bp; H+/K+ATPase α subunit-F,
5′-ACTCTGGGCTCCACGTCG-3′, H+/K+ATPase α
subunit-R, 5′-AGGATGGAGCTGCAGCGC-3′, 470 bp; UBCP-F,
5′-AGAAGAAGTCTTACACCACTC-3′, UBCP-R, 5′-GTAAGTCAGACAACATTTGCC-3′,
203 bp. For COX-2/β-actin, the PCR mixture was heated to 95°C for 5
min and amplification was performed for 35 cycles: denaturation at
95°C for 50 sec, annealing at 58°C for 60 sec and extension at 72°C
for 60 sec. Following the final cycle, the reactions were incubated
at 72°C for an additional 10 min. For
H+/K+ATPase α subunit/UBCP, amplification was
performed for 35 cycles: denaturation at 95°C for 60 sec, annealing
at 62°C for 60 sec and extension at 72°C for 60 sec. PCR products
were electrophoretically separated on 1.5% agarose gels containing
0.5 μg/ml ethidium bromide and visualized under ultraviolet
transillumination. Quantification of COX-2 and
H+/K+ATPase ATPase α subunit PCR products
were standardized in comparison with the housekeeping gene,
β-actin, and UBCP products, respectively, by densitometry. The mRNA
expression levels of the control group was expressed as 100%.
Levels of mRNA expression were presented as a percentage of the
control.
Analysis of protein expression by
FCM
Goat anti-COX-2 IgG antibody (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) was used as primary antibody
and FITC-labeled rabbit anti-goat IgG antibody was used as
secondary antibody for indirect immunofluorescence according to the
manufacturer’s instructions. Briefly, AGS and GES-1 cells, treated
with the vehicle or IL-1β (10 ng/ml) for the indicated times, were
washed with PBS and fixed in 4% paraformaldehyde for 40 min.
Following fixation, cells were washed twice and treated with 0.2%
Triton X-100 containing 5% FBS on ice for 10 min. Cells were washed
again with PBS and then incubated with a 1:50 dilution of
anti-COX-2 antibody at 4°C. Following incubation for 40 min, cells
were washed twice, further incubated with a 1:100 dilution of the
FITC-labeled secondary antibody at 4°C for 40 min, washed twice and
filtrated through a 50-μm nylon mesh. Specific fluorescence was
measured by FCM. For data acquisition, a gate was set on intact
cells by forward/side scatter analysis and a minimum of 10,000
events were analyzed. Protein expression levels are presented as
the mean fluorescence intensity (MFI) and expressed as the relative
MFI following correction for non-specific fluorescence using the
isotype control (MFICOX-2/MFIisotype
control).
Measurement of acid secretion
Intracellular accumulation of
14C-aminopyrine (14C-AP) was used as an
indirect measurement of functional acid secretory activity by
parietal cells (11,12). Cultured parietal cells, treated
with vehicle or IL-1β (10 ng/ml) for the indicated times, were
washed with EBSS containing 0.2% BSA, 2 mmol/l glutamine, 20 mmol/l
HEPES (pH 7.4) to remove dead and non-adherent cells and
resuspended in the medium described above at 1.0×106
cells/ml. 14C-AP (0.1 μCi; GE Healthcare Biosciences,
Pittsburgh, PA, USA) was added to 1.0 ml of the cell suspension and
the mixture was equilibrated at 37°C for 15 min. Following thorough
mixing of the cells, secretagogue histamine (10−4
mmol/l; Sigma-Aldrich) was added to stimulate the parietal cells to
uptake 14C-AP and the cells were incubated at 37°C for
30 min in an atmosphere of 5% CO2 and 95% air.
Incubation was terminated by rapidly removing the medium by
centrifugation and washing twice with EBSS solution. The cell
pellet was lysed with 0.5 ml 1% Triton X-100. Aliquots of cell
lysates were counted in a Beckman LS-6800 liquid scintillation
counter with dinitrophenol (DNP) correction. DNP (0.1 mmol/l) was
added separately to assess non-specific incorporation and values
were subtracted from the test and control values. 14C-AP
uptake was expressed as a percentage of control and calculated as:
(14C-AP uptake of IL-1β group - 14C-AP uptake
of DNP group)/(14C-AP uptake of control group -
14C-AP uptake of DNP group).
Statistical analysis
Values are expressed as the means ± SD of at least 6
independent experiments. Six replicates were performed in each
experiment. The Student’s t-test, one-way ANOVA and Mann-Whitney U
test were performed for statistical evaluation of the data using
the SPSS 17.0 statistical software package. P<0.05 was
considered to indicate a statistically significant difference.
Results
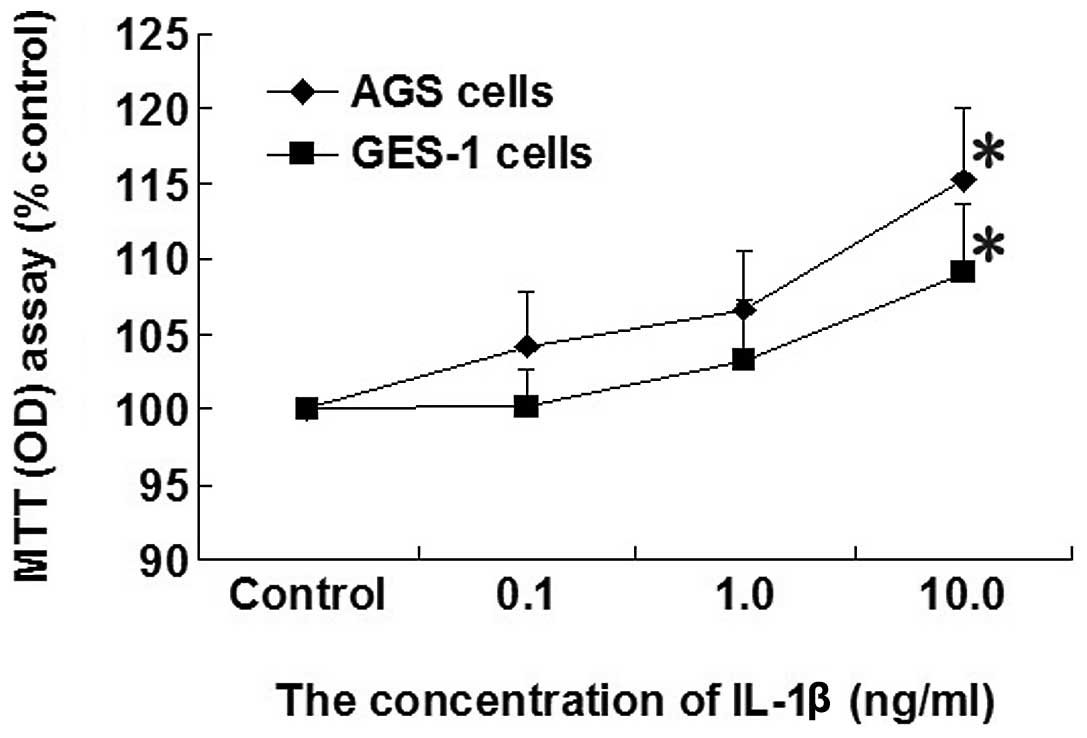
Effect of increasing concentrations of
IL-1β on cell proliferation in AGS and GES-1 cells
Cell proliferation was increased in AGS and GES-1
cells by increasing the concentration of IL-1β (0.1, 1.0 and 10
ng/ml). A significant increase in the proliferation rate in
response to IL-1β (10 ng/ml) was identified compared with the
control group in AGS and GES-1 cells (P<0.05; Fig. 1).
Effect of IL-1β on H. pylori-induced
apoptosis in AGS and GES-1 cells
The apoptosis of AGS and GES-1 cells was
significantly increased in the H. pylori group compared with
the control (P<0.05; Fig. 2).
IL-1β (10 ng/ml) exposure for 24 h significantly attenuated the
H. pylori-induced apoptosis by 61.1 and 56% in the AGS cells
and GES-1 cells, respectively, compared with the H. pylori
group (P<0.05; Fig. 2).
Effect of IL-1β on expression of COX-2
mRNA in AGS and GES-1 cells
Compared with the control group, mRNA expression of
COX-2 in the AGS cell line was significantly upregulated following
treatment with IL-1β (10 ng/ml) for 8 h (P<0.05; Fig. 3). A similar result was obtained in
the GES-1 cell line (P<0.05).
Effect of IL-1β on expression of COX-2
protein in AGS and GES-1 cells
As demonstrated in Fig.
4, the protein expression of COX-2 was significantly increased
following treatment with IL-1β (10 ng/ml) for 8 h in the AGS and
GES-1 cell lines compared with the control group (all P<0.05).
These results indicated that the effect of IL-1β on COX-2 protein
expression was consistent with the effect on COX-2 mRNA
expression.
Effect of IL-1β on acid secretion in
isolated rabbit parietal cells
The acid secretion stimulated by histamine
significantly inhibited by 14 and 50% following treatment with
IL-1β (10 ng/ml) for 30 min and 16 h, respectively, compared with
the control group in isolated rabbit parietal cells (all P<0.05;
Fig. 5). Compared with the group
treated with IL-1β (10 ng/ml) for 30 min, acid secretion stimulated
by histamine was significantly decreased in the group treated with
IL-1β (10 ng/ml) for 16 h. These results demonstrated that IL-1β
(10 ng/ml) inhibited acid secretion stimulated by histamine in a
time-dependent manner in isolated rabbit parietal cells.
Effect of IL-1β on
H+/K+ATPase α subunit mRNA expression in
isolated rabbit parietal cells
As demonstrated in Fig.
6, the mRNA expression of H+/K+ATPase α
subunit in isolated rabbit parietal cells was downregulated by 11
and 29% following treatment with IL-1β (10 ng/ml) for 30 min and 16
h, respectively, compared with the control group (all P<0.05).
Compared with the group treated with IL-1β (10 ng/ml) for 30 min,
the mRNA expression of the H+/K+ATPase α
subunit was significantly decreased in the group treated with IL-1β
(10 ng/ml) for 16 h. These results indicated that IL-1β (10 ng/ml)
downregulated the mRNA expression of
H+/K+ATPase α subunit in a time-dependent
manner in isolated rabbit parietal cells.
Discussion
Previous studies have demonstrated that human
gastric mucosa expresses COX-2 protein at low levels; however, the
expression of COX-2 is induced by H. pylori-associated
premalignant and malignant gastric lesions and correlates with the
depth of mucosal invasion, lymphatic invasion and metastasis in
human gastric carcinoma (11–13).
Specific and non-specific inhibitors of COX-2 suppressed
proliferation of cell lines that expressed high levels of COX-2.
However, these inhibitors exerted minimal effects on proliferation
of the cell lines expressing lower levels of COX-2 (14). In addition, COX-2 inhibitors
suppressed growth of gastric cancer xenografts by induction of
apoptosis and suppression of neoplastic cell replication (15). These results indicate that COX-2 is
important for the development of gastric cancer. The present study
identified the basal COX-2 expression in the transformed human
gastric cancer cell line (AGS) and human gastric epithelial cell
line (GES-1) using RT-PCR.
Although the mechanism of COX-2 regulation of cancer
development remains unclear, existing data indicate that COX-2
expression is associated with stimulation of cellular proliferation
and resistance to apoptosis. Previously, COX-2 inhibitor treatment
was observed to induce apoptosis, suppress cellular proliferation,
downregulate Bcl-2 expression and suppress the growth of
H-ras-transformed cells (16–18).
In addition, overexpression of COX-2 may induce expression of
epidermal growth factor receptor and metalloproteinase and decrease
expression of E-cadherin and transforming growth factor-β receptor
(15,19). These alterations are correlated
with enhanced tumorigenic potential and increased tumor
invasiveness.
COX-2 expression is associated with intensive
infiltration of inflammatory cells in H. pylori-infected
gastric mucosa where substantial amounts of cytokines are induced,
including IL-1β (20,21). A previous study by Zhang et
al demonstrated that H. pylori isogenic mutants
specifically lacking picA or picB, molecules responsible for
cytokine production in gastric cells, are less effective in
upregulation of COX-2 mRNA expression (22). These results indicate that picA and
picB may contribute to increased COX-2 expression and stimulation
of cytokine production. In the present study, the effect of
exogenous IL-1β on COX-2 expression of gastric epithelial cells was
analyzed. The results demonstrated that IL-1β induced the
expression of COX-2 mRNA and protein in GES-1 and AGS cell lines.
COX-2 expression induced by IL-1β may be mediated by activation of
multiple intracellular signaling pathways, including p44/42 and p38
MAPK, JNK and NF-κB (23,24). The results of the present study
revealed that IL-1β enhanced cellular proliferation and attenuated
H. pylori-induced apoptosis in GES-1 and AGS cell lines,
consistent with the observation that IL-1β induced expression of
COX-2. Therefore, we conclude that H. pylori-associated
IL-1β may stimulate cellular proliferation, inhibit H.
pylori-induced apoptosis and mediate gastric carcinogenesis
through upregulation of COX-2 expression. Additional mechanisms by
which IL-1β affects carcinogenesis may include IL-1β induction of
angiogenin mRNA and protein expression. Angiogenin is a
proangiogenic molecule associated with neovascularization of cancer
tissue. IL-1β may also induce carcinogenesis by stimulation of
metalloproteinases, thought to be important mediators of metastasis
(25,26). Maihöfner et al revealed that
expression of COX-2 was consistent with expression of IL-1β in
human colorectal cancer tissue, however, the association between
COX-2 and IL-1β in human gastric cancer tissue requires additional
investigation (27).
In humans, H. pylori infection may cause
acute epidemic gastritis associated with hypochlorhydria. In
certain individuals, chronic H. pylori infection causes
body-predominant gastritis and profound suppression of gastric acid
secretion that is partially reversible with eradication therapy
(28–30). The degree of acid suppression
depends on the distribution of H. pylori infection, scores
for activity and inflammation of gastritis in the body, the number
of H. pylori and the grade of colonization (31). In the present study,
14C-AP accumulation was performed to determine the
effects of exogenous IL-1β on acid secretion in isolated parietal
cells from rabbits. The results demonstrated that IL-1β exposure
for 30 min or 16 h inhibited histamine-stimulated acid secretion,
accompanied by downregulation of H+/K+ATPase
mRNA expression. The inhibitory potency of IL-1β was
time-dependent, as preincubation of parietal cells with IL-1β for
longer time intervals resulted in increased inhibition of
14C-AP accumulation and downregulation of
H+/K+ATPase mRNA expression. Since specific
H+/K+ATPase in parietal cell is the key
mediator of the final stages of gastric acid secretion, a decrease
in the level of H+/K+ATPase is hypothesized
to lead to a reduction of acid secretion (32). The present results indicate that
IL-1β may inhibit gastric acid secretion by downregulating
expression of H+/K+ATPase. However, the
possibility that IL-1β performs antisecretory functions in parietal
cells by blocking H+/K+ATPase activity cannot
be ruled out at present. Accumulating evidence indicates that the
inhibitory action of IL-1β is mediated by multiple intracellular
signaling pathways, including pertussis toxin and tyrosine
kinase-dependent and independent pathways (33,34).
Additional in vivo studies are consistent
with the present in vitro results. In Mongolian gerbils
inoculated orally with H. pylori for 6 and 12 weeks, serum
gastrin levels were increased and gastric acid output was
decreased. These alterations correlated with elevation of IL-1β
mRNA levels in gastric mucosa; however, gastric acid output and
serum gastrin level returned to control levels following
recombinant human IL-1 receptor antagonist (rhIL-1ra) injection. In
H. pylori-associated enlarged fold gastritis, increased
IL-1β release from gastric body mucosa was correlated with
decreased basal and tetragastrin-stimulated acid output, whereas
IL-1β release was significantly decreased with concomitant increase
in gastric acid secretion following eradication of H.
pylori. In patients infected with H. pylori, a
significant correlation was observed between IL-1β mRNA expression
in gastric fundic gland mucosa and gastric juice pH. Significant
decreases in the amount of IL-1β mRNA, gastric juice pH and serum
gastrin levels were observed in patients with eradication of H.
pylori, whereas no significant changes were observed in
patients without eradication (35). These results indicate that H.
pylori infection induces IL-1β expression and suppresses acid
secretion.
In addition to the direct effects on parietal cells,
the mechanisms by which the inhibitory effects of IL-1β on acid
secretion are mediated in vivo are: i) IL-1β induces
apoptosis in enterochromaffin-like cells (ECL cells) or inhibits
gastric histamine secretion and synthesis from ECL cells, leading
to the reduction in acid secretion stimulated by histamine
(36,37). ii) IL-1β mediates increased
prostaglandin E2 (PGE2) production via the
overexpression of COX-2. PGE2 functions as a potent
inhibitor of gastric acid secretion by directly retarding the
secretory function of parietal cells or reducing histamine release
from ECL cells. Moreover, PGE2 stimulates bicarbonate
secretion from gastric epithelial cells, which may contribute to a
decrease in gastric acidity (38–40).
A previous study in H. pylori-infected mice demonstrated
that increased PGE2 produced by overexpression of COX-2
stimulated cytokines (IL-1β) induced by H. pylori infection,
demonstrating the importance of PGE2 in gastric acid
hyposecretion by H. pylori infection (41).
One of the mechanisms involved in the development of
gastric cancer by H. pylori infection is associated with
long-term acid hyposecretion. Patients with hyposecretion are
exposed to hypergastrinemia, bacterial toxins, N-nitroso compounds
and products of inflammation, including reactive oxygen radicals
and nitrogen oxygen species, all well-known mutagens or carcinogens
(5,7). In hosts with low basal secretory
capacity and high IL-1β phenotypes, H. pylori is prone to
colonization of a wider niche involving the acid secretory corpus
region, resulting in higher levels of IL-1β production, resulting
in additional inhibition of acid secretion, a more aggressive body
gastritis and acceleration of gastric cancer development. A
previous study in Mongolian gerbils with low basal acid output and
genetic predisposition demonstrated that gerbils developed corpus
atrophy, intestinal metaplasia and were particularly prone to
developing gastric cancer when chronically colonized by H.
pylori infection (42). These
observations are consistent with a phenotype identified in human
individuals associated with increased risk of gastric cancer with
high IL-1β phenotypes.
In conclusion, the results from the present study
suggest that IL-1β may be a key mediator in H.
pylori-induced gastric carcinogenesis and a prime candidate as
a host genetic factor that may alter the risk of gastric cancer.
The present study demonstrates that IL-1β induced by H.
pylori infection is associated with the 2 mechanisms involved
in gastric carcinogenesis: i) IL-1β may promote cellular
proliferation, inhibit H. pylori-induced apoptosis by
upregulating COX-2 expression and lead to the disturbance of
gastric epithelial cell growth and ii) IL-1β may inhibit acid
secretion from parietal cells by downregulating
H+/K+ATPase expression.
Acknowledgements
The present study was supported and funded by the
National Natural Science Foundation of China (NSFC-30270600).
References
|
1
|
Momtaz H, Souod N, Dabiri H and Sarshar M:
Study of Helicobacter pylori genotype status in saliva,
dental plaques, stool and gastric biopsy samples. World J
Gastroenterol. 18:2105–2111. 2012.
|
|
2
|
Graham DY and Yamaoka Y: Disease-specific
Helicobacter pylori virulence factors: the unfulfilled
promise. Helicobacter. 5:3–9. 2000.
|
|
3
|
Hitzler I, Sayi A, Kohler E, et al:
Caspase-1 has both proinflammatory and regulatory properties in
Helicobacter infections, which are differentially mediated
by its substrates IL-1β and IL-18. J Immunol. 188:3594–3602.
2012.PubMed/NCBI
|
|
4
|
Albaker WI: Helicobacter pylori
infection and its relationship to metabolic syndrome: is it a myth
or fact? Saudi J Gastroenterol. 17:165–169. 2011. View Article : Google Scholar
|
|
5
|
El-Omar EM, Carrington M, Chow WH, et al:
Interleukin-1 polymorphisms associated with increased risk of
gastric cancer. Nature. 404:398–402. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tan IB, Ng I, Tai WM and Tan P:
Understanding the genetic basis of gastric cancer: recent advances.
Expert Rev Gastroenterol Hepatol. 6:335–341. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cai C and Zhu X: The Wnt/β-catenin pathway
regulates self-renewal of cancer stem-like cells in human gastric
cancer. Mol Med Report. 5:1191–1196. 2012.
|
|
8
|
Wang XC, Li Y, Fan LQ, et al: Integrase
interactor 1 regulates proliferation, apoptosis and invasion in
gastric cancer cells. Chin Med J (Engl). 125:527–532.
2012.PubMed/NCBI
|
|
9
|
Tu SP, Quante M, Bhagat G, et al: IFN-γ
inhibits gastric carcinogenesis by inducing epithelial cell
autophagy and T-cell apoptosis. Cancer Res. 71:4247–4259. 2011.
|
|
10
|
Li XB, Qian JM, Chen YJ and Chen YF:
Effect of lansoprazole on histamine-induced acid secretion in
isolated rabbit parietal cells. Chin J Dig Dis. 3:47–50. 2002.
View Article : Google Scholar
|
|
11
|
Sung JJ, Leung WK, Go MY, et al:
Cyclooxygenase-2 expression in Helicobacter
pylori-associated premalignant and malignant gastric lesions.
Am J Pathol. 157:729–735. 2000.
|
|
12
|
Pero R, Peluso S, Angrisano T, et al:
Chromatin and DNA methylation dynamics of Helicobacter
pylori-induced COX-2 activation. Int J Med Microbiol.
301:140–149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ohno R, Yoshinaga K, Fujita T, et al:
Depth of invasion parallels increased cyclooxygenase-2 levels in
patients with gastric carcinoma. Cancer. 91:1876–1881. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Y, Xu X and He P: Tubeimoside-1
inhibits proliferation and induces apoptosis by increasing the Bax
to Bcl-2 ratio and decreasing COX-2 expression in lung cancer A549
cells. Mol Med Report. 4:25–29. 2011.PubMed/NCBI
|
|
15
|
Gou HF, Chen XC, Zhu J, et al: Expressions
of COX-2 and VEGF-C in gastric cancer: correlations with
lymphangiogenesis and prognostic implications. J Exp Clin Cancer
Res. 30:142011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang DS, Shen KZ, Wei JF, et al: Specific
COX-2 inhibitor NS398 induces apoptosis in human liver cancer cell
line HepG2 through BCL-2. World J Gastroenterol. 11:204–207. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nandi J, Das PK, Zinkievich JM, et al:
Cyclo-oxygenase-1 inhibition increases acid secretion by modulating
H+,K+-ATPase expression and activation in
rabbit parietal cells. Clin Exp Pharmacol Physiol. 36:127–134.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Na HK, Inoue H and Surh YJ:
ET-18-O-CH3-induced apoptosis is causally linked to
COX-2 upregulation in H-ras transformed human breast epithelial
cells. FEBS Lett. 579:6279–6287. 2005.PubMed/NCBI
|
|
19
|
Timotheadou E, Skarlos DV, Samantas E, et
al: Evaluation of the prognostic role of a panel of biomarkers in
stage IB-IIIA non-small cell lung cancer patients. Anticancer Res.
27:4481–4489. 2007.PubMed/NCBI
|
|
20
|
Seo JH, Seo JY, Chung HY and Kim H: Effect
of pertussis toxin and herbimycin A on proteinase-activated
receptor 2-mediated cyclooxygenase 2 expression in Helicobacter
pylori-infected gastric epithelial AGS cells. Yonsei Med J.
52:522–526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tatsuguchi A, Sakamoto C, Wada K, et al:
Localisation of cyclooxygenase 1 and cyclooxygenase 2 in
Helicobacter pylori related gastritis and gastric ulcer
tissues in humans. Gut. 46:782–789. 2000.PubMed/NCBI
|
|
22
|
Zhang X, Zhong R, Zhang Z, et al:
Interaction of cyclooxygenase-2 promoter polymorphisms with
Helicobacter pylori infection and risk of gastric cancer.
Mol Carcinog. 50:876–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu W, Reinmuth N, Stoeltzing O, et al:
Cyclooxygenase-2 is up-regulated by interleukin-1 beta in human
colorectal cancer cells via multiple signaling pathways. Cancer
Res. 63:3632–3636. 2003.PubMed/NCBI
|
|
24
|
Catley MC, Chivers JE, Cambridge LM, et
al: IL-1beta-dependent activation of NF-kappaB mediates PGE2
release via the expression of cyclooxygenase-2 and microsomal
prostaglandin E synthase. FEBS Lett. 547:75–79. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Etoh T, Shibuta K, Barnard GF, et al:
Angiogenin expression in human colorectal cancer: the role of focal
macrophage infiltration. Clin Cancer Res. 6:3545–3551.
2000.PubMed/NCBI
|
|
26
|
Paduch R, Kandefer-Szerszeń M,
Szuster-Ciesielska A and Plewka K: Transforming growth factor-beta1
modulates metalloproteinase-2 and -9, nitric oxide, RhoA and
alpha-smooth muscle actin expression in colon adenocarcinoma cells.
Cell Biol Int. 34:213–223. 2010. View Article : Google Scholar
|
|
27
|
Maihöfner C, Charalambous MP, Bhambra U,
et al: Expression of cyclooxygenase-2 parallels expression of
interleukin-1beta, interleukin-6 and NF-kappaB in human colorectal
cancer. Carcinogenesis. 24:665–671. 2003.PubMed/NCBI
|
|
28
|
Sugimoto M, Furuta T, Shirai N, et al:
Evidence that the degree and duration of acid suppression are
related to Helicobacter pylori eradication by triple
therapy. Helicobacter. 12:317–323. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Oridate N, Takeda H, Yamamoto J, et al:
Helicobacter pylori seropositivity predicts outcomes of acid
suppression therapy for laryngopharyngeal reflux symptoms.
Laryngoscope. 116:547–553. 2006. View Article : Google Scholar
|
|
30
|
Zhu H, Hart CA, Sales D and Roberts NB:
Bacterial killing in gastric juice - effect of pH and pepsin on
Escherichia coli and Helicobacter pylori. J Med
Microbiol. 55:1265–1270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takashima M, Furuta T, Hanai H, et al:
Effects of Helicobacter pylori infection on gastric acid
secretion and serum gastrin levels in Mongolian gerbils. Gut.
48:765–773. 2001.
|
|
32
|
Nguyen NV, Gleeson PA, Courtois-Coutry N,
et al: Gastric parietal cell acid secretion in mice can be
regulated independently of H/K ATPase endocytosis.
Gastroenterology. 127:145–154. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wei XH, Yang T, Wu Q, et al: Peri-sciatic
administration of recombinant rat IL-1β induces mechanical
allodynia by activation of src-family kinases in spinal microglia
in rats. Exp Neurol. 234:389–397. 2012.
|
|
34
|
Shibata Y, Kasai H, Shimada M, et al:
IL-1β stimulates IL-8 production, including prostaglandin E2
receptor EP4-triggered pathways, in synoviocyte MH7A cells. Mol Med
Report. 2:359–363. 2009.
|
|
35
|
Sugimoto M, Furuta T and Yamaoka Y:
Influence of inflammatory cytokine polymorphisms on eradication
rates of Helicobacter pylori. J Gastroenterol Hepatol.
24:1725–1732. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kidd M, Gustafsson BI, Drozdov I and
Modlin IM: IL1beta- and LPS-induced serotonin secretion is
increased in EC cells derived from Crohn’s disease.
Neurogastroenterol Motil. 21:439–450. 2009.PubMed/NCBI
|
|
37
|
Beales IL: Gastrin and interleukin-1beta
stimulate growth factor secretion from cultured rabbit gastric
parietal cells. Life Sci. 75:2983–2995. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shimamoto C, Nakanishi Y, Katsu K, et al:
Prostaglandin E2 release in gastric antral mucosa of guinea-pigs:
basal PGE2 release by cyclo-oxygenase 2 and ACh-stimulated PGE2
release by cyclo-oxygenase 1. Exp Physiol. 91:1015–1024. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zinkievich JM, George S, Jha S, et al:
Gastric acid is the key modulator in the pathogenesis of
non-steroidal anti-inflammatory drug-induced ulceration in rats.
Clin Exp Pharmacol Physiol. 37:654–661. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sekiguchi F, Ohi A, Maeda Y, et al:
Delayed production of arachidonic acid contributes to the delay of
proteinase-activated receptor-1 (PAR1)-triggered prostaglandin E2
release in rat gastric epithelial RGM1 cells. J Cell Biochem.
112:909–915. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xiao F, Furuta T, Takashima M, et al:
Effects of cyclooxygenase-2 inhibitor on gastric acid secretion in
Helicobacter pylori-infected C57BL/6 mice. Scand J
Gastroenterol. 36:577–583. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pal J, Sanal MG and Gopal GJ: Vitamin-C as
anti-Helicobacter pylori agent: More prophylactic than
curative - Critical review. Indian J Pharmacol. 43:624–627.
2011.
|