Introduction
Hemorrhagic shock (HS), which is typically observed
following traumatic and surgical injuries, leads to organ
hypoperfusion, ischemia and additional reperfusion injuries during
resuscitation. HS and resuscitation (HSR) frequently induces
systemic inflammatory responses that result in lung inflammation
(the lung is the first targeted organ). Advanced phase HSR leads to
acute lung injury (ALI), which is responsible for the substantial
morbidity and mortality in critically ill patients (1,2).
Pathogenesis is characterized by increased production of
pro-inflammatory mediators and the activation of pulmonary
inflammatory cells, which leads to development of interstitial
edema and impaired lung function (3,4).
Therefore, modulation of these inflammatory responses may provide
an important avenue for therapeutic intervention when treating lung
injuries, including HSR-induced ALI.
Carbon monoxide (CO) is commonly considered
poisonous in high concentrations due to its ability to interfere
with oxygen delivery. However, CO is produced by the enzymatic
reaction of heme oxygenase-1 (a stress-inducible protein) in the
body and previous evidence indicates that endogenous CO is
important in the physiological functioning of organs and possesses
anti-inflammatory properties that have applications in a variety of
biomedical fields (5). Moreover,
administration of CO at low concentrations has been demonstrated to
mediate potent cytoprotective and anti-inflammatory effects during
hyperoxic lung injuries (6),
endotoxemia (7),
ischemic/reperfusion injuries (8)
and HSR-induced multiple organ failure (MOF) (9). We previously demonstrated that
inhalation of CO at low concentrations both prior to and following
HSR exerts a potent protective effect against ALI in rats, as
determined by improvements in histological findings as well as the
suppression of the inflammatory signaling cascade (10). However, to evaluate the possible
clinical applications of CO as a therapeutic modality, we must
determine the protective effects of CO at various stages, including
prior to or following resuscitation, as well as clarify the
molecular mechanisms behind the anti-inflammatory effects of
CO.
In the present study, we have demonstrated that CO
inhalation at a low concentration following resuscitation
ameliorates HSR-induced ALI by attenuating apoptotic cell death in
association with upregulation of peroxisome proliferator-activated
receptor (PPAR)-γ, an anti-inflammatory transcriptional regulator.
Present findings are likely to aid the expansion of new therapeutic
avenues for CO gas in the field of therapeutic medical gas and
critical care.
Materials and methods
Animals
The studies was approved by the Animal Use and Care
Committee of the Okayama University Medical School (Okayama, Japan)
in accordance with the guidelines for the care and use of
laboratory animals that follows the ARRIVE reporting guidelines
(11). Male Sprague-Dawley rats
weighing 380–420 g (Clea Japan, Inc., Tokyo, Japan) were housed in
a temperature-controlled (25°C) room with a 12-h light/dark cycle
and free access to food and water until the start of the
experiments.
HS protocol
All rats were subjected to sham or HS under
pentobarbital anesthesia (sodium pentobarbital: 50 mg/kg, i.p.) as
previously described (12).
Briefly, HS was initiated by withdrawing blood from the left
femoral vein to achieve a mean arterial blood pressure of 30±5 mmHg
which was maintained for 1 h. Arterial pressure was measured via
catheters inserted into the left femoral artery. Rats were
resuscitated with shed blood. The sham group underwent the same
instrumentation procedures, with the exception of bleeding.
Experimental design
Rats were randomly assigned to the following three
groups: sham-operated control (Sham), air-treated HSR (HSR/AIR) and
CO-treated HSR (HSR/CO). CO exposure was performed at 250 ppm for 3
h following the completion of resuscitation, as previously
described (10). Rats in the Sham
and HSR groups were maintained in room air in laminar flow cages.
Some HSR rats were exposed to CO at 250 ppm for 1 h prior to the
onset of HS (CO/HSR). At specific time points following
resuscitation, rats were sacrificed by decapitation under light
anesthesia with ethyl ether. The lungs were excised and frozen
immediately in liquid nitrogen and stored at −80°C until further
use.
In an additional set of experiments, HSR/AIR and
HSR/CO rats were subjected to a median laparotomy under ether
anesthesia for 3 h following the onset of resuscitation, in order
to examine the effect of CO exposure on the CO content of lung
tissues. Blood was collected from the abdominal aorta and the lungs
were perfused in situ with physiological saline until the
venous effluent became clear. The lungs were then removed for
preparation of tissue homogenates.
Histological study
Tissues fixed in 10% neutral-buffered formalin were
embedded in paraffin and cut in 4–6 μm-thick sections. After
deparaffinization, the sections were stained with hematoxylin and
eosin followed by a light microscope examination by a blinded
observer. Grading of lung injury severity was performed according
to previously described methods with modifications for five
independent experiments (13–15).
Lung sections adjacent to those used for
histopathological study were subjected to staining for neutrophils
with a naphthol AS-D chloroacetate esterase-staining kit (Sigma
Diagnostics, St. Louis, MO, USA) as described previously (16). The number of positively stained
cells was counted in five nonconsecutive sections per rat at a ×400
magnification by a blinded observer.
Lung wet weight to dry weight ratio
Lung tissue samples were blotted and weighed (wet
weight) before drying for 24 h at 110°C (dry weight). The wet
weight/dry weight ratio was calculated by dividing the wet weight
by the dry weight as an index of pulmonary edema (17).
RNA isolation and northern blot
analysis
Total RNA was isolated from the frozen rat tissues
with Tri-Reagent™ (Sigma) according to the manufacturer's
instructions. Northern blotting was performed as described
previously (17). Briefly, 20 μg
of total RNA was separated on a 1.2% (w/v) agarose gel containing
6.5% (v/v) formaldehyde and transferred on Bio-Rad Zeta-Probe
membrane (Bio-Rad Laboratories, Richmond, CA, USA). The blotted
membrane was hybridized with [α-32P]dCTP-labeled cDNA
probes for tumor necrosis factor (TNF)-α and inducible nitric oxide
synthase (iNOS), respectively, followed by washing at high
stringency. The membrane was exposed to X-ray films at −70°C, and
autoradiographs and 18S ribosomal RNA were quantified with an image
scanner (GelPrintTM 2000i, Genomic Solutions, Ann Arbor, MI, USA)
and computerized image analysis software (Basic Quantifier™ version
3.0, Genomic Solutions). The relative amounts of radiolabeled cDNA
that hybridized to the blots were normalized to 18S ribosomal RNA
levels for correction of loading errors.
Preparation of nuclear extracts and
western blot analysis
Nuclear extracts were isolated from the lung tissues
using Nuclear and Cytoplasmic Extraction Reagents (NE-PER™; Thermo
Fisher Scientific, Inc., Rockford, IL, USA), as previously
described (18). The nuclear
protein content was determined using the BCA Protein Assay kit™
(Thermo Fisher Scientific, Inc.). Samples were analyzed on a 12.5%
polyacrylamide gel (e-PAGEL™; ATTO, Tokyo, Japan), according to the
manufacturer's instructions. Following electrophoresis, proteins
were transferred to a Hybond-P PVDF membrane (GE Healthcare, Little
Chalfont, UK) and blocked with 4% (w/v) BlockAce™ (DS Pharma
Biomedical Co., Ltd., Osaka, Japan). Blots were incubated with
rabbit anti-PPAR-γ polyclonal antibody (1:1,200; Abcam, Cambridge,
UK) and then treated with horseradish peroxidase-labeled goat
anti-rabbit immunoglobulin G (IgG; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA). The antigen-antibody complexes were
visualized using an ECL™ chemiluminescence system (GE
Healthcare).
Real-time reverse transcription
(RT)-polymerase chain reaction (PCR)
Real-time RT-PCR was performed using SYBR Premix Ex
Taq™ (Takara Bio, Shiga, Japan) and a LightCycler (Roche
Diagnostics GmbH, Mannheim, Germany), as previously described
(16). The interleukin-10 (IL-10)
messenger RNA (mRNA) level was normalized to the β-actin mRNA
level.
Histological detection of apoptotic cell
death
Detection and quantification of apoptotic cells were
performed using the terminal deoxynucleotidyl transferase-mediated
dUTP-fluorescein isothiocyanate (FITC) nick-end labeling (TUNEL)
method with the MEBSTAIN Apoptosis kit II (MBL, Nagoya, Japan),
according to the manufacturer's instructions. Sections were
incubated with FITC-labeled avidin and then counterstained with 0.5
μg/ml propidium iodide to visualize the nuclei.
Immunostaining of activated caspase-3 was performed
using rabbit polyclonal primary antibody (1:250; Asp 175, no. 9661;
Cell Signaling Technology, Danvers, MA, USA), according to the
manufacturer's instructions. Sections were incubated with diluted
Cyanine 3-conjugated donkey anti-rabbit secondary antibody (1:100;
Chemicon International, Temecula, CA, USA).
Apoptotic and activated caspase-3-positive cells
were counted using a confocal laser scanning microscope (model
LSM510; ZEISS, Jena, Germany).
Measurement of carboxyhemoglobin (COHb)
and tissue CO content
Blood COHb levels in the arterial blood were
measured using the OSM3 Hemoximeter (Radiometer Copenhagen,
Copenhagen, Denmark). The tissue CO content was measured as
previously described (10).
Detection of tissue hypoxia by
pimonidazole adducts
Levels of tissue hypoxia were detected by
pimonidazole hydrochloride staining on lung (Hypoxyprobe™-1,
Natural Pharmacia International, Inc., Burlington, MA, USA) as
described previously (19). Slides
were stained with a primary mouse monoclonal antibody for
pimonidazole adducts, followed by a secondary FITC donkey
anti-mouse IgG (Chemicon International). Images were captured as
described earlier. Hypoxic rats breathed 6% oxygen continuously for
90 min as positive control. The atmosphere in the cages was sampled
continuously from the chamber exhaust outlet and the O2
concentration was monitored with a gas analyzer (CAPNOMAC™,
Datex-Ohmeda, Finland).
Statistical analysis
Statistical analysis was undertaken using the
Student's t-test or analysis of variance followed by Scheffé's
F-test, as appropriate. P<0.05 was considered to indicate a
statistically significant result. Data are shown as the mean ± SEM.
We used statistical package Statview (Abacus Concepts, Berkeley,
CA, USA).
Results
Therapeutic effect of CO inhalation on
HSR-induced lung injury
While sections obtained from the Sham rats appeared
normal, sections from HSR rats developed interstitial edema, as
demonstrated by pronounced alveolar septal thickening and
inflammatory cell infiltration 12 h after HSR (Fig. 1). CO exposure for 3 h following HSR
markedly mitigated these pathological changes, including
congestion, edema, inflammation and hemorrhage; however, CO
exposure for 1 h prior to HSR did not affect the pathological
findings (Fig. 1A). The
significant effects of CO exposure following HSR were also
confirmed by the scoring system based on histopathological changes
reviewed by a blinded independent researcher. Only CO inhalation
following HSR suppressed lung injuries, as revealed by histological
damage that was confirmed by a decrease in the total histological
score (Fig. 1B). By contrast, CO
inhalation prior to HSR did not affect the histopathological scores
(Fig. 1C).
 | Figure 1Comparative effects of carbon
monoxide (CO) exposure at various times on histological changes in
the lungs following hemorrhagic shock and resuscitation (HSR). (A)
Rats subjected to HSR were exposed to CO for 1 h prior to onset of
hemorrhagic shock or for 3 h following resuscitation or were
maintained in air. Lungs from HSR rats following these various
treatments were excised 12 h after resuscitation and subjected to
histological examination. Representative images from five
independent experiments (hematoxylin-eosin staining, ×200 original
magnification). (B) Severity of histopathological changes in the
lungs were graded according to criteria for congestion, edema,
inflammation and hemorrhage. Ten areas of the lung parenchyma from
each rat were graded as 0 (no findings or normal), 1 (mild), 2
(moderate) or 3 (severe) for each of the four parameters. (C) Sum
of histological scores for each of the four parameters were
calculated (n=5, for each group). *P<0.01 vs. Sham;
†P<0.05 vs. Sham; #P<0.01 vs. HSR/AIR;
¥P<0.01 vs. CO/HSR. Sham, sham-operated control rats;
HSR/AIR, HSR rats maintained in air; CO/HSR, HSR rats exposed to CO
for 1 h prior to hemorrhagic shock; HSR/CO, HSR rats exposed to CO
for 3 h following resuscitation |
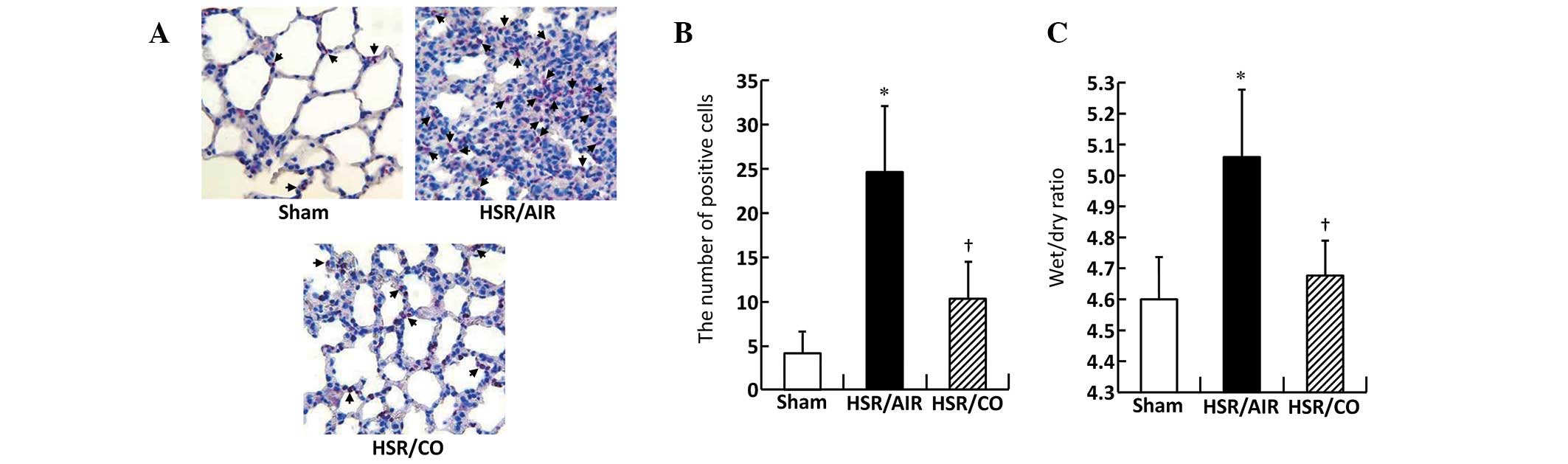
Effect of CO inhalation on neutrophil
sequestration in lungs following HSR
The number of infiltrating neutrophils was markedly
increased in the HSR/AIR group at 12 h following resuscitation
compared with that in the Sham group (Fig. 2A and B). By contrast, neutrophil
recruitment was identified as significantly reduced in the lungs of
the CO-treated rats (Fig. 2A and
B).
CO inhalation reduced lung edema
following HSR
Lung wet/dry ratio, a parameter of lung edema, was
identified as significantly increased within 12 h in the HSR group,
compared with that of the Sham group (Fig. 2C). However, CO inhalation following
HSR significantly attenuated HSR-induced lung edema (Fig. 2C).
Effect of CO treatment on the gene
expression of HSR-induced inflammatory mediators, including TNF-α
and iNOS
Although mRNA levels of TNF-α and iNOS were barely
detectable in the Sham group, these genes were significantly
upregulated in the HSR/AIR group at 3 h after resuscitation
(Fig. 3). CO inhalation following
HSR significantly decreased mRNA expression levels of TNF-α and
iNOS to almost the same levels as those measured in the Sham group
(Fig. 3).
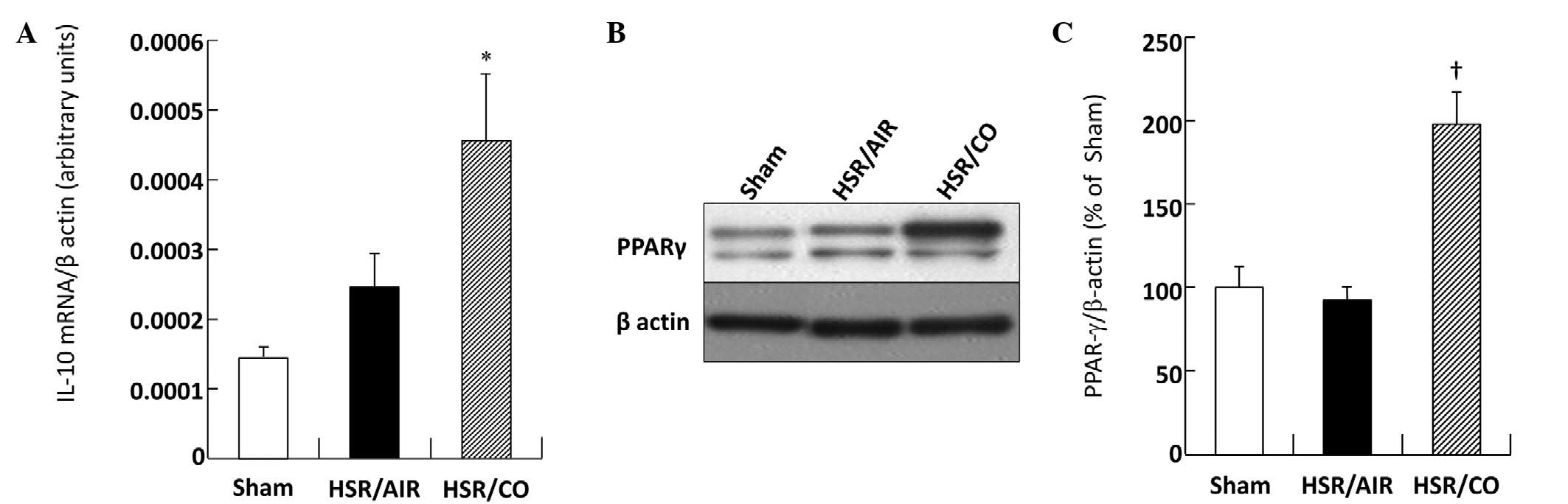
Effect of CO treatment on the expression
of IL-10 and PPAR-γ in the lungs following HSR
Pulmonary IL-10 mRNA was barely detectable in the
Sham group (Fig. 4A). The HSR
procedure slightly increased the IL-10 mRNA level in the HSR/AIR
group compared with that in the Sham group (Fig. 4A). Notably, CO treatment resulted
in further upregulation of IL-10 mRNA (Fig. 4A). While PPAR-γ protein was
expressed at high levels in the Sham group, levels in the HSR/AIR
group was almost the same as that in the Sham group at 3 h
following resuscitation (Fig. 4B and
C). However, CO treatment significantly increased this level in
comparison to that measured in the HSR/AIR group (Fig. 4B and C). The level measured in the
HSR/CO group was almost twice as large as that measured in the
HSR/AIR group (Fig. 4B and C).
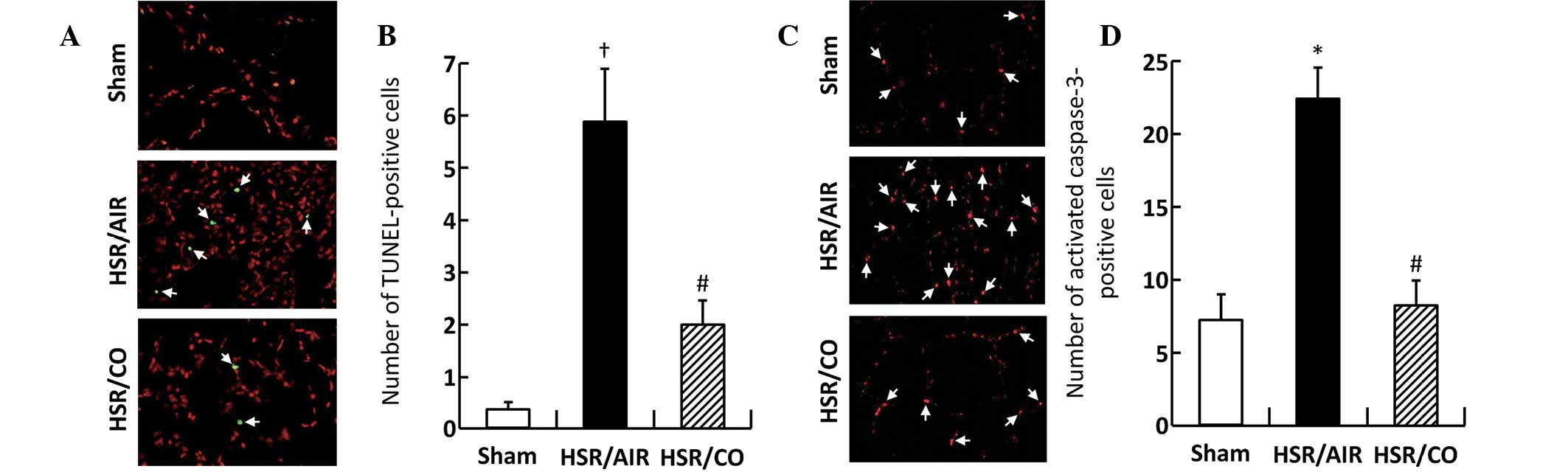
Effect of CO inhalation on apoptotic cell
death in the lungs
While TUNEL-positive cells were barely detected in
the Sham group, their numbers were significantly increased at 12 h
following HSR in the lungs of the experimental rats (Fig. 5A and B). By contrast, CO inhalation
significantly decreased the number by ~30% of the level measured in
the HSR/AIR group (Fig. 5A and B).
Activated caspase-3-positive cells were barely detected in the
lungs of the Sham group, while their number was significantly
increased at 12 h following resuscitation in the HSR/AIR rats
(Fig. 5C and D). By contrast, the
number of activated caspase-3-positive cells in the CO-treated HSR
rats was low low (P<0.027; HSR/AIR vs. HSR/CO) and at almost the
same level as that measured in the Sham group (Fig. 5C and D).
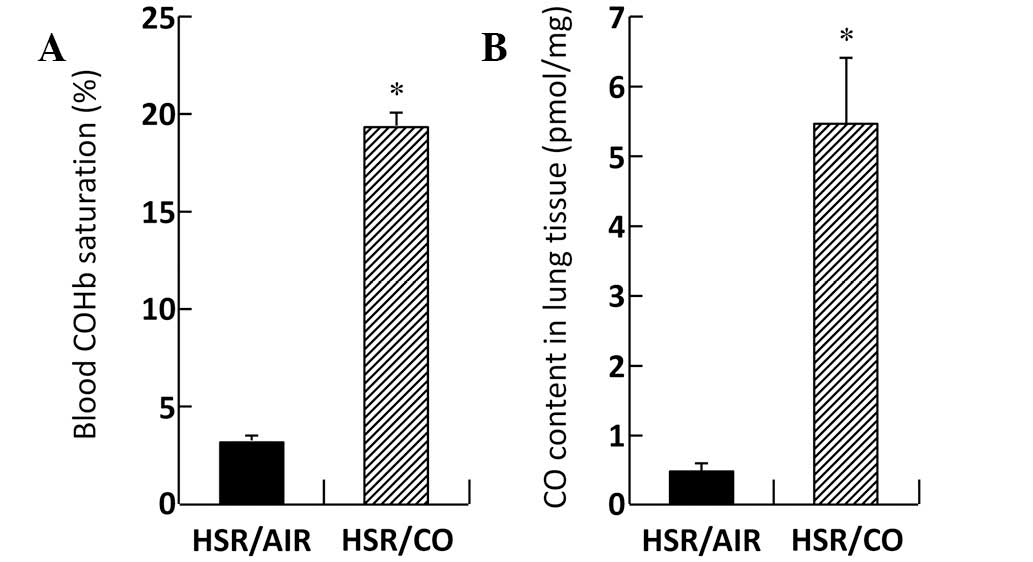
Effect of CO inhalation on blood COHb
levels and CO tissue content in the lungs
Blood COHb levels in the HSR/CO group were markedly
increased (19.40±0.66%) compared with those in the HSR/AIR group
(3.08±0.06%; Fig. 6A). CO content
in the lungs was markedly increased to 5.43±0.89 pmol/mg tissue
following CO exposure 3 h after resuscitation, while CO content in
the lungs of the HSR/AIR group was 0.49±0.02 pmol/mg tissue
(Fig. 6B).
CO did not affect the hemodynamic status
during HSR
Mean arterial pressure and heart rate in the HSR/CO
group were almost identical to those in the HSR/AIR group, even at
3 h after resuscitation (Table I).
Moreover, CO exposure did not exert a significant effect on pH,
partial pressure of oxygen (PO2) and partial pressure of
carbon dioxide (PCO2) levels in the arterial blood
(Table I).
 | Table IEffect of inhaled carbon monoxide
(CO) on central hemodynamics following HSR. |
Table I
Effect of inhaled carbon monoxide
(CO) on central hemodynamics following HSR.
| Mean arterial
pressure (mmHg) | Heart rate
(beats/min) | Arterial blood gas
analysis (3 h after HSR) |
|---|
|
|
|
|
|---|
| Rat treatment | End of
resuscitation | 3 h after HSR | End of
resuscitation | 3 h after HSR | pH | PO2
(mmHg) | PCO2
(mmHg) |
|---|
| HSR/AIR | 100.0±3.03 | 70.0±5.76 | 315.2±4.08 | 344.0±21.76 | 7.412±0.013 | 101.4±5.96 | 44.8±1.80 |
| HSR/CO | 100.4±10.97 | 73.2±8.38 | 284.8±18.00 | 297.6±5.31 | 7.412±0.013 | 117.0±12.95 | 40.7±1.36 |
CO did not induce tissue hypoxia
While there was no pimonidazole-binding in
sham-operated control rats, sections from unoperated animals
exposed to hypoxia, used as positive controls, revealed
significantly enhanced pimonidazole-binding compared with
sham-operated control rats (Fig.
7). By contrast, sections from HSR rats 3 h after resuscitation
revealed no pimonidazole-binding, regardless of CO inhalation
(Fig. 7).
Discussion
The present study demonstrates that inhalation of CO
at a low concentration following HSR exerts potent therapeutic
effects against HSR-induced ALI. The findings of this study
indicate that CO treatment suppresses molecular activation of
HSR-induced inflammatory events. CO exerted anti-inflammatory
effects that are, at least in part, mediated through induction of
IL-10, an anti-inflammatory cytokine and activation of PPAR-γ, an
anti-inflammatory transcriptional regulator. HS leads to
exaggerated systemic inflammatory responses, which may be important
in the development of MOF. MOF is associated with high mortality
rates, reported in specific cases to exceed 50% (1). ALI is the first clinical
manifestation of organ failure and a major factor leading to MOF,
which is characterized by acute lung inflammation involving the
local recruitment and activation of polymorphonuclear leukocytes
and the release of pro-inflammatory mediators (1,20).
Therefore, a novel anti-inflammatory modality that attenuates the
overwhelming lung inflammation that develops following HS-induced
lung injury may hold important therapeutic potential.
We previously demonstrated that combined treatment
of CO prior to and following HSR exerts potent anti-inflammatory
effects, resulting in reduced inflammatory cell influx into the
lungs and marked attenuation in the expression of pro-inflammatory
cytokines (10). The present study
demonstrates that exposure to CO alone following HSR confers
cytoprotective effects on HSR-induced lung injuries, primarily
through its anti-inflammatory effects. This finding suggests that
post-treatment use of CO may have potential as a therapeutic
approach against lung injuries and inflammation that result from
multiple causes. Prevention of lung injuries by treatment with CO
prior to noxious stimuli, including lipopolysaccharide (LPS),
hyperoxia and ischemia/reperfusion, have been described by other
investigators using experimental animal models (6,7,21).
The results of these studies are consistent with our findings, but
treatment with CO following harmful insult also has been reported
to provide protection against lung injuries that are induced by
various oxidative stimuli (22–25).
Novel observations of the present study provide further evidence
that post-treatment use of CO represents a potent therapeutic
modality for treatment of ALI, which may contribute to clinical
benefits in the treatment of lung injuries and inflammation.
CO possesses various anti-inflammatory properties
that contribute to the reduction of oxidative stress and apoptotic
cell death and are, at least in part, mediated through the p38
mitogen-activated protein kinase signaling pathway (26). However, the exact molecular
mechanisms of anti-inflammatory actions of CO remain elusive.
Previous studies, including ours, indicate that CO inhibits the
activation of nuclear factor κB and activator protein-1, leading to
amelioration of inflammatory lung disease by various causes,
including LPS and HSR (10,27,28).
The beneficial effects of CO have also been reported in association
with the upregulation of IL-10 expression (26). Moreover, a previous study indicated
that the anti-inflammatory effects of CO involve the inhibition of
upstream toll-like receptor signaling pathways (29). Thus, multiple mechanistic actions
of CO have been postulated. Previous observations have suggested
another mechanism of CO that is exerted on the inflammatory
response, which is mediated by PPAR-γ (30,31).
In accordance with these reports, upregulation of PPAR-γ was
accompanied by the protective effects of CO against ALI following
HSR in the present study. Previous observations clearly demonstrate
that the endogenous ligands of PPAR-γ reduce organ injury and
dysfunction, including detrimental effects to the liver, kidneys,
intestines and lungs, which are caused by HS (32). More recently, augmented PPAR-γ
expression by exogenous agonists was demonstrated to contribute to
the modulation of systemic and pulmonary inflammation in HSR
(33). These investigators also
indicated that PPAR-γ agonists exert anti-apoptotic effects in the
lungs following severe hemorrhage (34). Further studies using
PPAR-γ-neutralizing antibodies or PPAR-γ-knockout animals are
likely to be important in clarification of the function of PPAR-γ
in HSR.
Although it is important to establish a clinically
applicable strategy for using CO in treatment of ALI following HSR,
CO exposure is associated with serious health problems. CO is well
known for its high affinity for hemoglobin, which is more than 200
times greater than that of oxygen, as well as its ability to block
the binding of oxygen (35),
thereby leading to impaired delivery of oxygen to tissues and
ultimately resulting in hypoxic tissue damage and death (36). In the present study, rats exposed
to 250 ppm CO for 3 h exhibited considerably high blood COHb levels
(19.40±0.66%). However, arterial blood gas analysis revealed that
there was no difference in PaO2 levels between CO and
AIR treated-rats in the HSR groups (Table I). Moreover, as shown in Fig. 7, CO inhalation did not induce lung
tissue hypoxia as determined by the pomonidazole hydrochloride
staining, which is known as an in vivo hypoxia marker
(19). Instead, CO treatment of
rats markedly improved the clinical presentation of HSR-induced
ALI.
There are two concerns regarding species
differences, particularly between small and large animals, prior to
clinical application of CO. One is the differences is rate of CO
uptake into the body. Longer reaction time is required to form COHb
in large animals, including humans, than in small animals,
including mice and rats, which is attributable to the lower minute
volume of ventilation/unit of body weight in large animals
(37). The other is differences in
affinity of CO to hemoglobin. The affinity of CO to hemoglobin is
approximately 4 times higher than that of oxygen in rodents
(38). The magnitude of the
affinity differences between two gases is significantly smaller in
rodents than in humans. Thus, humans may inhale higher
concentration of CO than mice to obtain the same level of CO in the
tissue which is released from COHb. However, it was recently
reported that CO inhalation only at 100 ppm suppresses LPS-induced
pulmonary inflammation in mice (39). This allows us to hypothesize that
CO exposure below the lethal toxicity dose may exert a protective
effect on lung injury and inflammation in the clinical setting.
In conclusion, the present study demonstrates that
HSR causes significant pulmonary inflammation, as shown by the
increase in the gene expression levels of inflammatory mediators,
neutrophil emigration and pulmonary edema, which leads to apoptotic
cell death. CO inhalation following HSR significantly reduces these
indices with additional increases in gene expression of IL-10, an
anti-inflammatory cytokine and protein expression of PPAR-γ, an
anti-inflammatory transcriptional regulator, resulting in
amelioration of HSR-induced injury. These findings indicate that CO
inhalation possesses a potent therapeutic effect on HSR-induced
lung injury, at least in part, through the attenuation of the
inflammatory signaling pathway. Although the application of CO to
therapeutic modalities are likely to require additional preclinical
and clinical studies on its safety and efficacy for human use,
strategies for the attenuation of the HSR-induced inflammatory
signaling pathway, as shown in the present study, may expand the
currently limited therapeutic options.
Acknowledgements
The authors thank Dr Reiko Akagi for providing cDNAs
for TNF-α and iNOS.
Abbreviations:
|
ALI
|
acute lung injury
|
|
CO
|
carbon monoxide
|
|
COHb
|
carboxyhemoglobin
|
|
FITC
|
fluorescein isothiocyanate
|
|
HS
|
hemorrhagic shock
|
|
HSR
|
hemorrhagic shock and
resuscitation
|
|
IgG
|
immunoglobulin G
|
|
IL-10
|
interleukin-10
|
|
iNOS
|
inducible nitric oxide synthase
|
|
LPS
|
lipopolysaccharide
|
|
MOF
|
multiple organ failure
|
|
mRNA
|
messenger ribonucleic acid
|
|
PCO2
|
partial pressure of carbon dioxide
|
|
PO2
|
partial pressure of oxygen
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
|
TNF
|
tumor necrosis factor
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP-FITC nick-end labeling
|
References
|
1
|
Bhatia M and Moochhala S: Role of
inflammatory mediators in the pathophysiology of acute respiratory
distress syndrome. J Pathol. 202:145–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hudson LD, Milberg JA, Anardi D and
Maunder RJ: Clinical risks for development of the acute respiratory
distress syndrome. Am J Respir Crit Care Med. 151:293–301. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kollef MH and Schuster DP: The acute
respiratory distress syndrome. N Eng J Med. 332:27–37. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bernard GR, Artigas A, Brigham KL, et al:
The American-European Consensus Conference on ARDS. Definitions,
mechanisms, relevant outcomes, and clinical trial coordination. Am
J Respir Crit Care Med. 149:818–824. 1994. View Article : Google Scholar
|
|
5
|
Foresti R, Bani-Hani MG and Motterlini R:
Use of carbon monoxide as a therapeutic agent: promises and
challenges. Intensive Care Med. 34:649–658. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Otterbein LE, Mantell LL and Choi AM:
Carbon monoxide provides protection against hyperoxic lung injury.
Am J Physiol. 276:L688–L694. 1999.PubMed/NCBI
|
|
7
|
Sarady JK, Zuckerbraum BS, Bilban M, et
al: Carbon monoxide protection against endotoxic shock involves
reciprocal effects on iNOS in the lung and liver. FASEB J.
18:854–856. 2004.PubMed/NCBI
|
|
8
|
Zhang X, Shan P, Otterbein LE, et al:
Carbon monoxide inhibition of apoptosis during ischemia-reperfusion
lung injury is dependent on the p38 mitogen-activated protein
kinase pathway and involves caspase 3. J Biol Chem. 278:1248–1258.
2003. View Article : Google Scholar
|
|
9
|
Zuckerbraun BS, McCloskey CA, Gallo D, Liu
F, Ifedigbo E, Otterbein LE and Billiar TR: Carbon monoxide
prevents multiple organ injury in a model of hemorrhagic shock and
resuscitation. Shock. 23:527–532. 2005.PubMed/NCBI
|
|
10
|
Kanagawa F, Takahashi T, Inoue K, et al:
Protective effect of carbon monoxide inhalation on lung injury
following hemorrhagic shock/resuscitation in rats. J Trauma.
69:185–194. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kikenny C, Browne WJ, Cuthill IC, Emerson
M and Aitman DG: Improving bioscience research reporting: The
ARRIVE guidelines for reporting animal research. J Pharmacol
Pharmacother. 1:94–99. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Inoue K, Takahashi T, Uehara K, et al:
Protective role of heme oxygenase 1 in the intestinal tissue injury
in hemorrhagic shock in rats. Shock. 29:252–261. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murakami K, McGuire R, Cox RA, et al:
Heparin nebulization attenuates acute lung injury in sepsis
following smoke inhalation in sheep. Shock. 18:236–241. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zegdi R, Fabre O, Cambillau M, et al:
Exhaled nitric oxide and acute lung injury in a rat model of
extracorporeal circulation. Shock. 20:569–574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang H, Meng F, Li W, Tong L, Qiao H and
Sun X: Splenectomy ameliorates acute multiple organ damage induced
by liver warm ischemia reperfusion in rats. Surgery. 141:32–40.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Umeda K, Takahashi T, Inoue K, et al:
Prevention of hemorrhagic shock-induced intestinal tissue injury by
glutamine via heme oxygenase-1 induction. Shock. 31:40–49. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maeshima K, Takahashi T, Uehara K, et al:
Prevention of hemorrhagic shock-induced lung injury by heme
arginate treatment in rats. Biochem Pharmacol. 69:1667–1680. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sunil VR, Patel KJ, Nilsen-Hamilton M,
Heck DE, Laskin JD and Laskin DL: Acute endotoxiemia is associated
with upregulation of lipocalin 24p3/Lcn2 in lung and liver. Exp Mol
Pathol. 83:177–187. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Faleo G, Neto JS, Kohmoto J, et al: Carbon
monoxide ameliorates renal cold ischemia-reperfusion injury with an
upregulation of vascular endothelial growth factor by activation of
hypoxia-inducible factor. Transplantation. 85:1833–1840. 2008.
View Article : Google Scholar
|
|
20
|
Ciesla DJ, Moore EE, Johnson JL, Burch JM,
Cothren CC and Sauaia A: The role of the lung in postinjury
multiple organ failure. Surgery. 138:749–757. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kohmoto J, Nakao A, Stolz DB, et al:
Carbon monoxide protects rat lung transplants from
ischemia-reperfusion injury via a mechanism involving p38 MAPK
pathway. Am J Transplant. 7:2279–2290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Song R, Kubo M, Morse D, et al: Carbon
monoxide induces cytoprotection in rat orthotopic lung
transplantation via anti-inflammatory and anti-apoptotic effects.
Am J Pathol. 163:231–242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zuckerbraun BS, Chin BY, Wegiel B, et al:
Carbon monoxide reverses established pulmonary hypertension. J Exp
Med. 203:2109–2119. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nemzek JA, Fry C and Abatan O: Low-dose
carbon monoxide treatment attenuates early pulmonary neutrophil
recruitment following acid aspiration. Am J Physiol Lung Cell Mol
Physiol. 294:L644–L653. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goebel U, Siepe M, Schwer CI, et al:
Postconditioning of the lungs with inhaled carbon monoxide
following cardiopulmonary bypass in pigs. Anesth Analg.
112:282–291. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Otterbein LE, Bach FH, Alam J, et al:
Carbon monoxide has anti-inflammatory effects involving the
mitogen-activated protein kinase pathway. Nat Med. 6:422–428.
2006.PubMed/NCBI
|
|
27
|
Sarady JK, Otterbein SL, Liu F, Otterbein
LE and Choi AM: Carbon monoxide modulates endotoxin-induced
production of granulocyte macrophage colony-stimulating factor in
macrophages. Am J Respir Cell Mol Biol. 27:739–745. 2002.
View Article : Google Scholar
|
|
28
|
Morse D, Pischke SE, Zhou Z, et al:
Suppression of inflammatory cytokine production by carbon monoxide
involves the JNK pathway and AP-1. J Biol Chem. 278:36993–36998.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakahira K, Kim HP, Geng XH, et al: Carbon
monoxide differentially inhibits TLR signaling pathways by
regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp
Med. 203:2377–2389. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bilban M, Bach FH, Otterbein SL, et al:
Carbon monoxide orchestrates a protective response through
PPARgamma. Immunity. 24:601–610. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hoetzel A, Dolinary T, Vallbracht S, et
al: Carbon monoxide protects against ventilator-induced via
PPAR-gamma and inhibition of Egr-1. Am J Respir Crit Care Med.
177:1223–1232. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abdelrahman M, Collin M and Thiemermann C:
The peroxisome proliferator-activated receptor-gamma ligand
15-deoxyDelta 12,24 prostaglandin J2 reduces the organ injury in
hemorrhagic shock. Shock. 22:555–561. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chima RS, Hake PW, Piraino G, Mangeshkar
P, Denenberg A and Zingarelli B: Ciglitazone ameliorates lung
inflammation by modulating the increased inhibitor kappaB protein
kinase/nuclear factor-kappaB pathway following hemorrhagic shock.
Crit Care Med. 36:2849–2857. 2008. View Article : Google Scholar
|
|
34
|
Chima RS, Hake PW, Piraino G, Mangeshkar
P, O'Connor M and Zingarelli B: Ciglitazone, a novel inhibitor of
lung apoptosis following hemorrhagic shock. Int J Clin Exp Med.
3:1–9. 2010.PubMed/NCBI
|
|
35
|
Rodkey FL, O'Neal JD and Collison HA:
Oxygen and carbon monoxide equilibria of human adult hemoglobin at
atmospheric and elevated pressure. Blood. 33:57–65. 1969.PubMed/NCBI
|
|
36
|
Morse D, Sethi J and Choi AM: Carbon
monoxide-dependent signaling. Crit Care Med. 30:S12–S17. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mayr FB, Spiel A, Leitner J, et al:
Effects of carbon monoxide inhalation during experimental
endotoxemia in humans. Am J Respir Crit Care Med. 171:354–360.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brittain T, Sutherland J and Greenwood C:
A study of the kinetics of the reaction of ligands with the
liganded states of mouse embryonic haemoglobins. Biochem J.
234:151–155. 1986.PubMed/NCBI
|
|
39
|
Wilson MR, O'Dea KP, Dorr AD, Yamamoto H,
Goddard ME and Takata M: Efficacy and safety of inhaled carbon
monoxide during pulmonary inflammation in mice. PLoS One.
5:e115652010. View Article : Google Scholar : PubMed/NCBI
|