Introduction
Mitotane, also known as o,p'-DDD or
(RS)-1-chloro-2-[2,2-dichloro-1-(4-chlorophenyl)-ethyl]-benzene,
was introduced 50 years ago and is currently the only FDA-approved
drug for the treatment of adrenocortical carcinoma (ACC) (1,2).
Plasma mitotane concentration in patients with ACC reaches 20 mg/l
(3). Plasma concentrations
exceeding this are associated with a number of side effects of the
digestive and central nervous systems (4). Mitotane therapy leads to a decrease
in the secretion of steroid hormone and therefore supplementary
glucocorticoid or mineralocorticoid therapy is recommended
(5). Mitotane has been shown to
increase patient survial in only 33% of cases (6) and studies have been performed to
analyze the efficacy of combinations of etoposide, doxorubicin,
cisplatin and mitotane or streptozocin and mitotane (7). A number of hypotheses have been put
forward to explain this narrow window of mitotane efficiency.
Concentration of mitotane appears to vary considerably between
patients due to differences in transport from the digestive system
to the blood stream and efficiency of liver detoxification
(7). A mitotane concentration
<14 mg/l has no effect in the majority of patients, therefore,
blood concentration is a limiting factor of mitotane efficiency
(8).
Frequent chromosomal abnormalities in ACCs may
affect the response of the adrenal gland to mitotane treatment.
Malignant aberrations have been reported in loci 17p (TP53)
and 11p15 (insulin-like growth factor-II) only (9,10).
Therefore, the identification of a suitable biomarker for the
prediction of ACC response to mitotane remains unsuccessful.
However, a biomarker to distinguish between adrenocortical adenoma
and ACC has been found by genome and transcriptome screening
(9,10). At present, the determination of
markers to predict the efficacy of mitotane therapy requires
additional studies.
There are two general applications of mitotane. The
first is for treatment of ACCs that cannot be surgically removed
and the second is as an adjuvant treatment following adrenalectomy
to prevent recurrence (7). A
biomarker for prediction of mitotane response is required for ACCs
that cannot be removed surgically as access to adrenal tissue is
not possible. The ideal biomarker of mitotane response would
measure a property of blood samples extracted from preoperative
patients. ACC tumor tissues obtained following surgery may be
diagnosed using a number of methods, including gene expression and
genetic aberration assays in the cells cultured in vitro.
Biomarkers must be extremely sensitive to a low dose of mitotane
and measured easily in a cell extract or culture.
Mitotane lowers secretion of steroid hormones by
targeting enzymes involved in the steroidogenic pathway, including
11β-hydroxylase [encoded by cytochrome P450 (CYP11) B1] and
cytochrome P450 side chain cleavage (P450scc, encoded by
CYP11A1) (5). Previously,
mitotane and its derivatives were analyzed in the NCI-H295R human
ACC cell line. Mitotane was reported to decrease cortisol and
aldosterone secretion as well as 3β-hydroxysteroid
dehydrogenase/δ5-4 isomerase type I (HSD3B1) mRNA levels
(11). By contrast, inconsistent
results were observed for the inhibitory effect of mitotane on the
expression of steroidogenic genes, steroidogenic acute regulatory
protein (StAR), CYP11B1, CYP17A1 and
CYP11B2 (gene-encoding aldosterone synthase) (11,12).
Adrenocortical cells secrete mineralocorticoids
(mainly aldosterone), glucocorticoids (mainly cortisol) and adrenal
androgens [androstendione and dehydroepiandrosterone sulfate
(DHEAS)] in the glomerulosa, fasciculata and reticularis zones of
the adrenal gland, respectively. The main substrate of
steroidogenesis is extracellular and intracellular cholesterol,
which is transported to the external mitochondrial membrane. StAR
transports cholesterol from the outer to the inner membrane of the
mitochondria. The first mitochondrial step of steroid hormone
synthesis is cleavage of cholesterol into pregnenolone by
cholesterol desmolaze (P450scc, EC 1.14.15.6, encoded by
CYP11A1) (13). Cholesterol
desmolaze, whose gene expression is regulated by
adrenocorticotropic hormone (ACTH) via cAMP and the PKA pathway,
limits synthesis of steroid hormones. The adrenal
17α-hydroxylase/17,20-lyase microsomal enzyme system includes
cytochrome P450 oxidoreductase (POR), cytochrome b5A
(CYB5A) and cytochrome P450c17 (steroid 17α-monooxygenase, encoded
by P450c17, EC 1.14.99.9, encoded by CYP17A1) (14,15).
CYP17A1 expression is stimulated by ACTH-triggered pathways
(16).
In addition to the inhibition of steroidogenesis,
mitotane acts as a cytotoxic drug (12,17),
hypothosized to be mediated by induction of apoptosis (18). However, the molecular mechanisms of
mitotane-mediated regulation of steroidogenesis and adrenocortical
cell viability remain unclear. Previous studies on the effect of
mitotane on the expression of genes encoding proteins involved in
steroid hormone production are contradictory (11,12).
In addition, cell viability results differ between these studies.
To the best of our knowledge, no studies have analyzed the
expression of genes associated with cell cycle regulation,
including cyclin dependent kinase inhibitor 1A, (encoding p21;
CDKN1A) and MYC (encoding cMyc), as well as genes
involved in important regulatory pathways in adrenocortical cells,
including transforming growth factor β1 (TGFβ1) and the PKA
regulatory subunit, PRKAR1A, following mitotane treatment.
In the present study, whether mitotane regulates the expression of
key genes involved in steroidogenesis, CYP11A1 and
CYP17A1, as well as important regulators of the cell cycle,
TGFB1, PRKAR1A, CDKN1A and MYC was
investigated. Since the human adrenocortical cell line NCI-H295R
originates from human ACC, this cell line was used to investigate
mitotane effects on cancerous adrenocortical cells. These genes
were selected following analysis of microarray data from publically
available resources (19) and from
previous studies describing genetic aberrations in ACC.
CYP11A1, CYP17A1, TGFB1, PRKAR1A and
MYC are located in loci in which aberrations have been
previously detected in ACC tumors (20–22).
CDKN1A is not located in a locus known to be aberrated in
ACC tumors. The present study is likely to aid selection of a
biomarker for distinguishing between mitotane-sensitive and
-insensitive ACCs.
Materials and methods
Materials
The human adrenocortical NCI-H295R cell line was
obtained from Dr W. Rainey (Dallas, TX, USA). Plasmids containing
the CYP11A1 promoter fragment (605 bp) were obtained from Dr
W. Miller (San Francisco, CA, USA) (23) and vectors containing the
CYP17A1 promoter fragments were obtained from Dr M. Sewer
(Nashville, TN, USA) (24).
Mitotane was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
NCI-H295R cells were cultured at 37°C and 5%
CO2/95% air in DMEM/F-12 (1:1 v/v) supplemented with
1.25% L-glutamine, 2.5% NuSerum (BD Biosciences, Franklin Lakes,
NJ, USA), 1% ITS plus (BD Biosciences) and antibiotic antimycotic
solution (Sigma-Aldrich). Confluent cells were incubated for 24 h
in DMEM/F-12 prior to treatment with mitotane.
Hormonal test
Following 24-h incubation with mitotane,
concentrations of cortisol and DHEAS in the media were measured
using an electrochemiluminescence immunoassay (Cobas 6000, Roche
Diagnostics, Burgess Hill, UK). Hormone concentrations were
normalized against total protein in the same well, determined by
the Bradford colorimetric assay (Bio-Rad, Hercules, CA, USA).
Cell viability and caspase-3 and −7
activities
To analyze cell viability, the Cell Growth
Determination kit,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich), was used. Following 24-h mitotane incubation, cells
(in 24-well plates) were incubated in DMEM/F-12 and 10% MTT (0.5
mg/ml) for 4 h. Media were removed and a solvent was added (0.1 M
hydrochloric acid in isopropanol). Following gentle mixing,
absorbance was measured at 630 nm and the background absorbance was
measured at 405 nm using a Stat-Fax 2100 spectrophotometer
(Awareness Technology, Inc., Palm City, FL, USA).
Caspase-3 and −7 activities were analyzed with the
Caspase-Glo 3/7 assay kit (Promega, Madison, WI, USA). After 24-h
stimulation, the Caspase-Glo 3/7 reagent was added to the cell
culture medium in a 96-well plate in a 1:4 ratio and incubated for
1 h. Luminescence was measured using a TD-20/20 luminometer (Turner
BioSystems, Sunnyvale, CA, USA).
Gene expression
Following incubation, total RNA was extracted from
the cultured cells using TRItidy G, according to the manufacturer's
instructions (Applichem, Darmstadt, Germany). Extracted total RNA
(1 μg) was reverse-transcribed using the Superscript Reverse
Transcriptase kit (Life Technologies, Carlsbad, CA, USA).
Oligo(dT)15-primed cDNAs were amplified by quantitative PCR (qPCR)
using the primers listed in Table
I and the LightCycler FastStart DNA Master SYBR-Green I kit
(Roche Diagnostics). Crossing points were calculated automatically
based on a second derivative algorithm and the results were
analyzed by the relative expression method. mRNA levels of
hydroxymethylbilane synthase (HMBS) and 39S ribosomal
protein L19, mitochondrial (MRPL19) were determined and used
as reference genes.
 | Table IOligonucleotide sequences used in
quantitative PCR. |
Table I
Oligonucleotide sequences used in
quantitative PCR.
| Gene name | Oligonucleotide
sequence | Amplicon length
(bp) | ENSEMBL accession
number |
|---|
| MYC |
5′-CCTACCCTCTCAACGAC-3′ | | |
|
5′-ATCTTCTTGTTCCTCCTCAG-3′ | 188 |
ENSG00000136997 |
| CYP11A1 |
5′-TGTTGAAGAAGTCGGCAG-3′ | | |
|
5′-TAGTGATGGACTCAAAGG-3′ | 216 |
ENST00000268053 |
| CYP17A1 |
5′-TGGCCCCATCTATTCTGTTC-3′ | | |
|
5′-CTTCTCCAGCTTCTGATCGC-3′ | 454 |
ENSG00000148795 |
| HMBS |
5′-GCCAAGGACCAGGACATC-3′ | | |
|
5′-TCAGGTACAGTTGCCCATC-3′ | 160 |
ENST00000442944 |
| MRPL19 |
5′-ACTTTATAATCCTCGGGTC-3′ | | |
|
5′-ACTTTCAGCTCATTAACAG-3′ | 171 |
ENST00000393909 |
| CDKN1A |
5′-CCAGCATGACAGATTTCTAC-3′ | | |
|
5′-CACACAAACTGAGACTAAGG-3′ | 148 |
ENST00000244741 |
| PRKAR1A |
5′-TCCTCATGGGAAGCACAC-3′ | | |
|
5′-AGCTGACCCCTCTAAAATAA-3′ | 200 |
ENST00000358598 |
| TGFB1 |
5′-GAAACCCACAACGAAATC-3′ | | |
|
5′-AATTTCCCCTCCACGGCT-3′ | 300 |
ENST00000358598 |
Transient transfection and luciferase
assay
Following culture, cells were cotransfected, using
Lipofectamine PLUS (Invitrogen Life Technologies, Carlsbad, CA,
USA), with pGL3 plasmid containing −57, −300 and −700 bp fragments
of the CYP17A1 promoter or a −605 bp fragment of the
CYP11A1 promoter (0.5 μg) fused to Photinus
luciferase and the pRL-TK plasmid (0.05 μg) containing the
Renilla luciferase gene, the latter of which was used as a
control of transfection efficiency.
Following transfection, cells were incubated for 24
h with mitotane, harvested and lysed with passive lysis buffer.
Luciferase activities in the cell lysates were determined using the
Dual Luciferase System (Promega) and TD-20/20 luminometer (Turner
BioSystems).
Statistical analysis
Results (mean ± SEM) from at least three independent
experiments were expressed as the fold change from a control value.
One-way analysis of variance was also used to evaluate the results
obtained by qPCR. Post-hoc comparisons were performed with the
Student-Newman-Keuls test. P<0.05 was considered to indicate a
statistically significant difference.
Results
The effect of mitotane on NCI-H295R cell
viability and activities of caspase-3 and −7
Mitotane is a potent inhibitor of adrenocortical
cancer cell growth. NCI-H295R adrenocortical cells were incubated
with various concentrations of mitotane for 24 h. Concentrations
between 0 and 2 mg/l (6.25 μM) did not affect NCI-H295R viability
(Fig. 1A). However, 20 mg/l (62.5
μM) mitotane caused a 20% decrease in cell viability (Fig. 1A). Since blood concentration of
mitotane has been identified to be ~14 mg/l (43.75 μM) in ACC
patients and is detected for considerably longer than 24 h in the
blood, mitotane may markedly decrease viability of ACC cells.
 | Figure 1NCI-H295R cell viability and
caspase-3 and −7 activity in cells treated with various doses of
mitotane. (A) NCI-H295R cells were incubated with 0.2, 2.0 and 20
mg/l (0.625, 6.25 and 62.5 μM) mitotane for 24 h followed by
incubation with MTT for 4 h. (B) NCI-H295R cells were incubated
with 0.2, 2.0, 4.0, 8.0, 16, 20 and 32 mg/l (0.625, 6.25, 12.5, 25,
50, 62.5 and 100 μM) mitotane to measure caspase-3 and −7 activity.
Following 24 h, cells were incubated with a caspase detection
buffer and the luminescent signal was measured following 1 h. Data
are presented as the percentage of the control (100%). Each
experiment was repeated four times, error bars represent ±SEM.
**P<0.01, ***P<0.001. |
Mitotane concentrations between 0.2 (0.6 μM) and 16
mg/l (50 μM) did not cause a detectable change in activities of
caspase-3 or −7 (Fig. 1B).
Mitotane doses between 20 (62.5 μM) and 32 mg/l (100 μM) led to an
increase in caspase-3 and −7 activities by up to a 2.0- and
2.5-fold, respectively. These data indicate that inhibition of
NCI-H295R cell viability by mitotane may be caused by
apoptosis.
Regulation of steroid hormone secretion
by mitotane in NCI-H295R cells
Low doses of mitotane, between 2 (6.25 μM) and 4
mg/l (12.5 μM), were sufficient to cause an 80% decrease of
cortisol and DHEAS secretion (Fig.
2). Higher doses of mitotane between 8 (25 μM) and 32 mg/l (100
μM) did not lead to greater inhibition of cortisol secretion.
However, DHEAS secretion was diminished by 90 and 95% in cells
treated with 8 (25 μM) and 32 mg/l (100 μM) mitotane,
respectively.
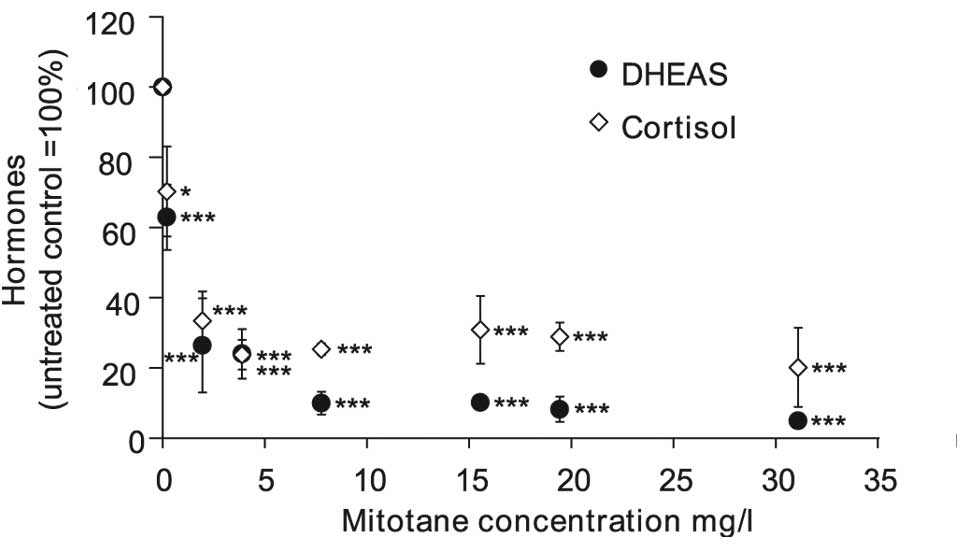 | Figure 2Cortisol and DHEAS secretion in
NCI-H295R cells treated with various doses of mitotane. NCI-H295R
cells were incubated with 0.2, 2.0, 4.0, 8.0, 16, 20 and 32 mg/l
(0.625, 6.25, 12.5, 25, 50, 62.5 and 100 μM) mitotane. Following 24
h, cortisol and DHEAS levels were measured by ECLIA. Data are
presented as the percentage of the control (100%). Each experiment
was repeated four times, error bars represent ±SEM.
*P<0.05, ***P<0.001. ECLIA,
electrochemiluminescence immunoassay. DHEAS, dehydroepiandrosterone
sulfate. |
The mean concentration of cortisol in the media was
18.5 nM for control and 2.55 nM in 20 mg/l (62.5 μM) mitotane
following 24-h incubation. Cortisol concentrations in the media
were calculated as pmol/μg protein. Control was 0.45 pmol/μg and 20
mg/l mitotane was 0.13 pmol/μg following 24-h incubation. Data
shown in Fig. 2 represent pmol/μg
values transformed into percentage of control (100%).
The mean concentration of DHEAS in the media was 440
and 130 nM for control and 20 mg/l (62.5 μM) mitotane,
respectively. Following calculation of DHEAS concentration in the
media as pmol/μg protein, control was 6.3 pmol/μg and 20 mg/l (62.5
μM) mitotane was 0.47 pmol/μg.
The effect of mitotane on gene expression
in NCI-H295R cells
To verify which genes are regulated in NCI-H295R
cells, mRNA levels of CYP11A1, CYP17A1,
CDKN1A, MYC, TGFB1 and PRKAR1A were
determined in NCI-H295R cells treated with 0.2, 2.0 and 20 mg/l
mitotane for 24 h (Fig. 3). mRNA
encoding enzymatic proteins involved in steroidogenesis,
CYP11A1 and CYP17A1, were decreased by 72 and 65%,
respectively, in cells treated with 20 mg/l mitotane. CDKN1A
and MYC mRNA levels were not affected by mitotane.
TGFB1 and PRKAR1A mRNA levels were reduced by 65 and
57%, respectively, by 20 mg/l (62.5 μM) mitotane.
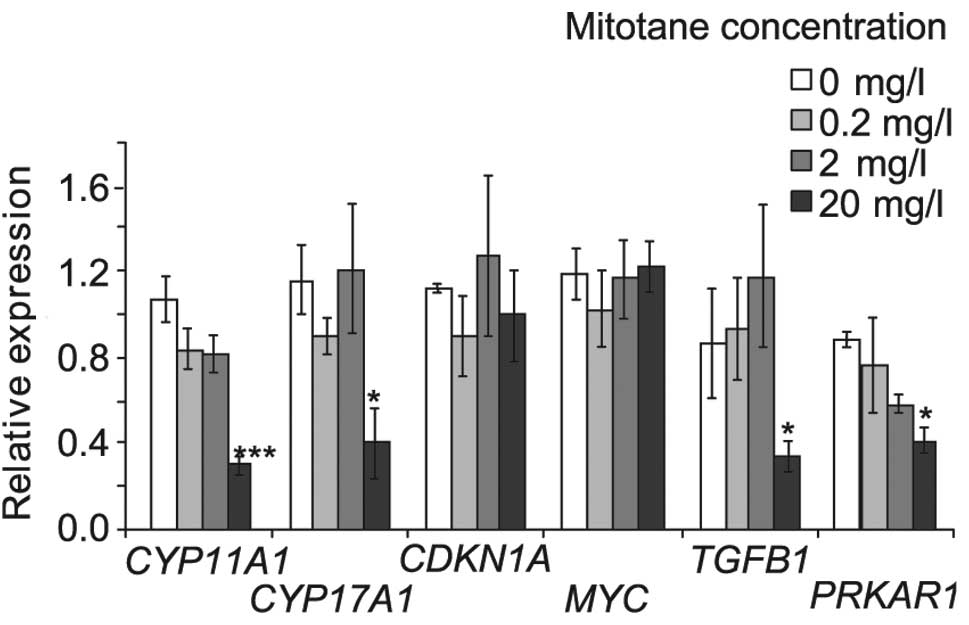 | Figure 3mRNA levels of CYP11A1,
CYP17A1, CDKN1A, MYC, TGFB1 and
PRKAR1A in NCI-H295R cells treated with 0.2, 2.0 and 20 mg/l
(0.625, 6.25 and 62.5 μM) mitotane for 24 h. MRPL19 and
HMBS genes were used as internal references. mRNA levels
were measured by qPCR. Data were analyzed by the relative
expression method and each experiment was repeated four times.
Error bars represent ±SEM. *P<0.05,
**P<0.01, ***P<0.001. CYP,
cytochrome P450; PRKAR1A, protein kinase A regulatory
subunit; TGFB1, transforming growth factor β. |
In addition, mRNA expression levels of genes
encoding enzymes involved in catalyzing limiting steps of
steroidogenesis were analyzed to determine the mechanism of
mitotane inhibition of steroidogenesis. Incubation of NCI-H295R
cells with between 0.2 and 32 mg/l mitotane reduced CYP11A1
and CYP17A1 mRNA levels (Fig.
4).
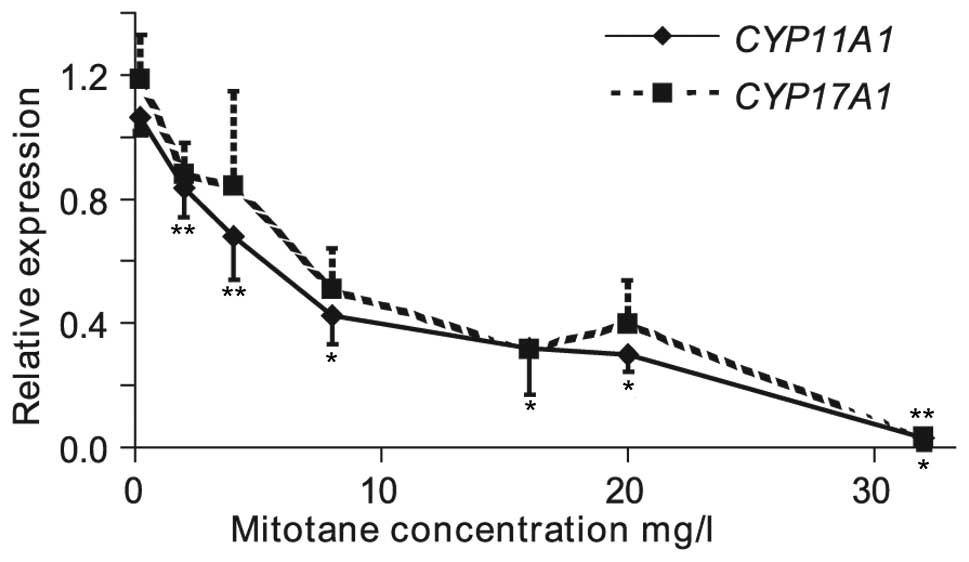 | Figure 4mRNA levels of CYP11A1 and
CYP17A1 in NCI-H295R cells treated with 0.2, 2.0, 4.0, 8.0,
16, 20 and 32 mg/l (0.625, 6.25, 12.5, 25, 50, 62.5 and 100 μM)
mitotane for 24 h. mRNA levels were measured by qPCR. MRPL19
and HMBS mRNA levels were used as internal references. Data
were analyzed by the relative expression method and each experiment
was repeated at least three times. Error bars represent ±SEM, only
down bars corresponding to -SEM are included for CYP11A1 and
only up bars corresponding to +SEM are included for CYP17A1.
*P<0.001, **P<0.05. |
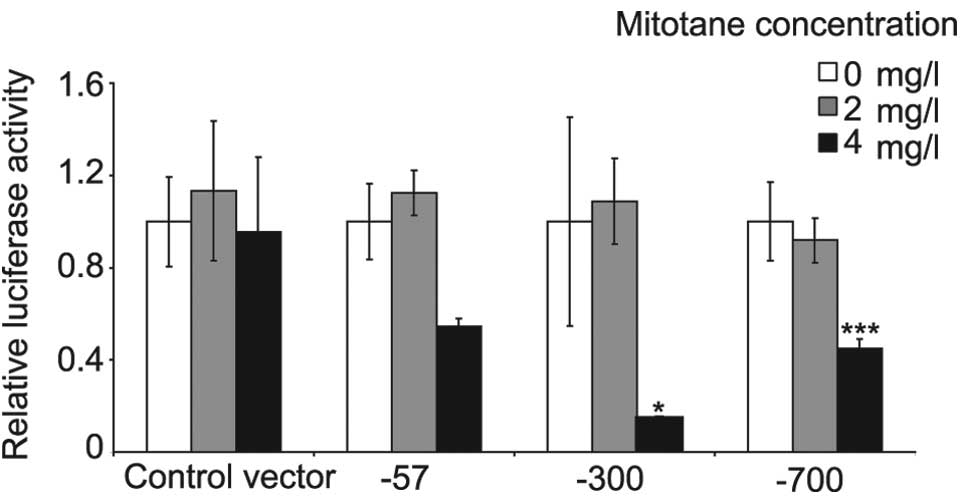
Mitotane inhibits CYP17A1 promoter
activity
To determine whether mitotane targets CYP17A1
expression at the transcriptional or post-transcriptional level,
NCI-H295R cells were transfected with plasmids containing 3 (57,
300 or 700 bp) upstream fragments of the CYP17A1 promoter
fused to the luciferase gene. The fragments of CYP17A1 were
inhibited by 4 mg/l mitotane following 24-h incubation. No effect
was observed at 2 mg/l mitotane. The transfection system was
compromised by >4 mg/l mitotane and the luciferase activity
signal was not detectable. Significant inhibition of the 300 and
700 bp fragments by mitotane was observed. These fragments contain
cAMP response elements indicating that CYP17A1 promoter
regulation by cAMP and mitotane may be coupled (Fig. 5).
Discussion
Mitotane, an anti-ACC drug has been known for a
number of years to reduce adrenocortical steroidogenesis and cell
growth (17). Previously, mitotane
and its derivatives were revealed to decrease the viability of Y1
and NCI-H295R cells (11,25) as well as steroidogenesis in
NCI-H295R cells (11,12,25).
The present study is consistent with results of Lin et al
demonstrating that mitotane decreased expression of CYP11A1
and CYP17A1 genes in NCI-H295R cells. In addition, current
results reveal that activity of the CYP17A1 promoter was
diminished by mitotane (12). For
the first time decreased viability of NCI-H295R cells was
accompanied by increased caspase-3 and −7 activities. Expression of
MYC and CDKN1A, two important genes involved in cell
cycle regulation, was unchanged. By contrast, TGFB1 and
PRKAR1A mRNA levels were diminished.
Previously, Asp et al observed decreased
viability of NCI-H295R cells in the presence of mitotane (11). However, a 90% reduction in cell
viability was noted in NCI-H295R cells treated for 72 h with 6.4
mg/l (20 μM) mitotane compared with 20% in the present study. Lin
et al did not observe a decrease in cell viability with 12.8
mg/l (40 μM) mitotane (12). The
highest concentration of mitotane used in the current study was 20
mg/l (62.5 μM), which was found to decrease cell viability. This
concentration is the highest level of mitotane achieved in the
plasma of patients treated against ACC. These inconsistencies in
results between studies may be caused by variations in cell culture
conditions or genotype variants of NCI-H295R cell lines originating
from ACC. At present these results make it difficult to predict the
effect of mitotane in vivo. However, genetic variability in
NCI-H295R cells may be considered to represent in vivo
conditions where genetic diversity in ACCs result in variable
outcomes of mitotane treatment in ACC patients (2).
Reduced cell survival following mitotane treatment
may be caused by decreased cell viability or the number of cell
divisions as well as cell cycle arrest or apoptosis. Similar to
mitotane, 1,1-dichloro-2,2 bis (p-chlorophenyl) ethylene (DDE) is a
DDT derivative and has been demonstrated to induce apoptosis in rat
Sertoli cells. In addition, DDE has been observed to increase
caspase-3 mRNA levels (26).
However, a previous study in MCF-7 estrogen responsive cells,
demonstrated that estrogenic organochlorine pesticides, including
o,p'-DDT, mimic the endogenous estrogen, 17β-estradiol and suppress
apoptosis (27). To determine the
mechanism by which mitotane reduces cell number, caspase-3 and −7
activities were determined. Caspase-3 and −7 are executioner
caspases present in the cytosol as inactive zymogen dimers which
are activated by initiator caspases-8 and −9 (28). Lakhani et al previously
reported that caspase-3 and −7 regulate mitochondrial events in the
apoptotic pathway (29). Caspase-3
and −7 exhibit analogous properties and demonstrate similar
substrate specificities (30,31).
It has been hypothesized that caspase-3 activation does not
necessarily lead to terminal apoptosis. DNA electrophoresis
following 24-h mitotane treatment was performed (data not shown and
a typical apoptotic ladder of DNA bands was not observed,
inconsistent with the apoptotic effect of mitotane) (32,33).
Further studies to determine whether mitotane induces programmed
cell death or necrosis must be performed.
TGFβ1 regulates adrenal development, inhibits
adrenal steroid production and decreases cell proliferation,
therefore, in the present study TGFB1 gene expression was
investigated (34,35). The growth factor alters the cell
cycle, activates caspase-3 and induces apoptosis in adrenocortical
cells via small mothers against decapentaplegic (SMAD) proteins
(36). Decreased expression of
TGFB1 and concomitant stable expression of CDKN1A
were observed as well as increased caspase-3 activity. Therefore,
TGFβ1 was not considered to be a key factor in the mechanism of
mitotane and the drug was hypothesized to trigger caspase-3
activity independently of the TGFβ1/SMAD/p21 pathway. Mitotane was
observed to have similar effects in NCI-H295R as aspirin in human
umbilical vein endothelial cells, suppressing TGFβ1 without
altering expression of CDKN1A(37).
To determine whether decreased viability in
NCI-H295R cell was mediated by an increase in execution caspases, a
mediator of the mitotane effect, other than TGFβ1, which is
involved in cell cycle regulation was investigated. MYC and
CDKN1A were hypothesized to be involved in this process.
cMyc is an coordinator of cell proliferation and apoptosis,
repressing specific survival pathways that regulate caspases. This
process may be the mechanism by which cMyc promotes death
receptor-induced apoptosis (38).
cMyc affects a number of cell cycle regulators inducing
caspase-dependent and -independent apoptosis (39). MYC expression is inhibited
by TGFβ1 which triggers apoptosis in NCI-H295R cells (36). MYC mRNA expression levels
were unchanged by mitotane, therefore we concluded that cMyc is not
involved in mitotane dependent apoptosis in NCI-H295R cells or the
decrease of viability is not a result of apoptosis. A previous
study reported that p,p'-DDE may induce testicular apoptosis in
rats through mitochondrial pathways, releasing cytochrome c and
additional proapoptotic factors to activate caspase-3 and −7, which
later promote cell death by directly processing and activating
caspase-8 (26).
The cyclin-dependent kinase inhibitor, p21, is
activated by p53-dependent and -independent mechanisms following
stress, leading to cell cycle arrest and under specific conditions,
induction of apoptosis (40).
CDKN1A gene expression was analyzed to determine
mitotane-induced cytotoxicity. Expression of CDKN1A was not
affected by mitotane. In general, CDKN1A expression is
upregulated by apoptosis stimulating drugs, however, there are
exceptions, such as sodium butyrate, an inhibitor of histone
deacetylases (41). Sodium
butyrate treatment increases caspase-3 expression in human gastric
cancer cells, however, the expression of TP53 and
CDKN1A is unchanged (41).
Alterations in the expression of CDKN1A is
not the only mechanism by which p21 activity is regulated.
Phosphorylation of p21 by PKA leads to the formation of
procaspase-3/p21 complexes rendering human hepatoma HepG2 cells
resistant to Fas-mediated apoptosis (42). In the presence of mitotane, a
decreased expression of PRKAR1A was observed. Therefore, we
hypothesized that a low expression of PRKAR1A enables the
free catalytic subunit of PKA to phosphorylate p21, blocking
subsequent procaspase-3 cleavage. Inconsistent with this
hypothesis, caspase-3 activity in the current study was increased,
indicating that although inactivation of procaspase-3 by
phosphorylated p21 blocked apoptosis in HepG2 cells, this mechanism
does not occur in NCI-H295R cells. If the pathway is presumed to be
active and catalytic subunit of PKA phosphorylates p21, the gradual
decrease of ATP production by damaged mitochondria following
treatment with mitotane may be responsible for the rupture of the
procaspase-3/p21 complex. Mitochondrial damage begins with
mitochondrial outer membrane permeabilization leading to the
release of cytochrome c, activation of caspase-3 and −7 and then
loss of transmembrane potential and collapse of ATP synthesis.
These mitochondrial events may contribute to the failure of
steroidogenesis (43).
A gradual decrease of cortisol and DHEAS in
NCI-H295R cells following mitotane treatment was observed. Asp
et al previously identified a comparable effect on cortisol
secretion in NCI-H295R cells (11). Cortisol secretion was maximally
inhibited by 4 mg/l mitotane and DHEAS was maximally inhibited by 8
mg/l mitotane. Since these low doses of mitotane decreased cortisol
secretion, we hypothesized that steroid hormone production is
interrupted by additional mechanisms to mitochondria damage.
o,p'-DDT derivatives may partially mimic the activity of endogenous
estrogen in MCF-7 cells (44). In
NCI-H295R cells, estradiol alone increases production of DHEAS. By
contrast, non-steroidal synthetic estrogen diethylstilbestrol does
not stimulate DHEAS production. Estradiol and diethylstilbestrol
inhibited the NCI-H295R production of cortisol (45). The antisteroidogenic effect of
mitotane on NCI-H295R is different to the effect of estrogen on
these cells, therefore the anticancer drug and sex steroid were
hypothesized to function via different pathways. Activin, a member
of the TGFβ1 family, inhibits steroidogenesis in NCI-H295R cells
decreasing the secretion of cortisol and DHEAS as well as promoting
apoptosis, however, this non-selective effect, similar to mitotane,
is mediated by SMAD proteins (46).
NCI-H295R cells may be a suitable model for
identification of a biomarker of mitotane efficacy. Collection and
selection of various mitotane- and non-resistant strains of
NCI-H295R cells for genome and transcriptome analysis of sensitive
and non-sensitive cells must be performed in further studies to
identify a biomarker.
In the present study, a mitotane-mediated decrease
in the expression of key steroidogenic genes, CYP11A1 and
CYP17A1, was detected. The effect of mitotane on
CYP11A1 and CYP17A1 expression levels was analyzed
using a wider range of mitotane concentrations between 0.2 and 32
mg/l, including 20 mg/l, the maximal concentration of mitotane
detected in patients treated for ACC. CYP11A1 and
CYP17A1 are involved in cortisol and DHEAS synthesis. The
proteins encoded by these genes are P450 cytochromes which function
in the mitochondria and endoplasmic reticulum of adrenocortical
cells. CYP11A1 and CYP17A1 mRNA levels gradually
declined as the concentration of mitotane increased. By contrast, a
stimulatory effect on CYP17A1 expression was previously
observed by Asp et al in NCI-H295R cells treated with 1.6
mg/l mitotane (11). However, the
authors did not report effects of higher concentrations of the
drug. More recently, Lin et al reported results on mitotane
treatment on CYP11A1 and CYP17A1 mRNA levels,
consistent with the current study (12).
In this study, analysis of CYP11A1 promoter
fragments indicated that CYP11A1 is not regulated by
mitotane at the transcriptional level (data not shown). By
contrast, the promoter activity of CYP17A1 is markedly
inhibited by mitotane. Mitotane inhibited the activity of the −300
bp CYP17A1 promoter fragment, comprising a cAMP response
element, indicating that activation by cAMP and inhibition by
mitotane may be associated with common mechanisms (24).
The impairment of cortisol and DHEAS secretion by
mitotane may be explained by reduced CYP17A1 and
CYP11A1 gene expression in NCI-H295R cells treated with the
drug. In addition, the toxic effect of mitotane on mitochondria may
also explain abnormalities in steroid synthesis. Mitochondria may
also be involved in reduced cell viability promoted by mitotane.
Increased caspase-3 and −7 activities indicate that the reduced
number of cells following mitotane treatment may be caused by
apoptosis. Further investigations are required to explain the
mechanism behind the effects of mitotane on cell viability, since
the drug did not affect cMyc or p21, two key cell cycle regulators
and an apoptotic DNA ladder was not observed. CYP11A1,
CYP17A1, TGFB1 and PRKAR1A were hypothesized
to be candidates for biomarkers of the mitotane effect since mRNA
expression of these genes were altered by treatment with mitotane.
In addition, caspase or transfection analysis of the CYP17A1
promoter must be performed. Therefore, to analyze these biomarkers,
patient cell culture must be performed and treated with mitotane
for subsequent gene expression analysis and caspase response
assays. Epigenome, genome aberration and transcriptome analysis
must also be performed.
Acknowledgements
The authors thank Beata Raczak and Bogumiła
Ratajczak for help during preparation of the current study. This
study was partially supported by grants from the Polish Ministry of
Science and Higher Education (N N 403 598538) and the Poznan
University of Medical Sciences (no. 501-01-1124182-07635.)
References
|
1
|
Hahner S and Fassnacht M: Mitotane for
adrenocortical carcinoma treatment. Curr Opin Investig Drugs.
6:386–394. 2005.PubMed/NCBI
|
|
2
|
Huang H and Fojo T: Adjuvant mitotane for
adrenocortical cancer - a recurring controversy. J Clin Endocrinol
Metab. 93:3730–3732. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hermsen IG, Fassnacht M, Terzolo M,
Houterman S, den Hartigh J, Leboulleux S, Daffara F, Berruti A,
Chadarevian R, Schlumberger M, Allolio B, Haak HR and Baudin E:
Plasma concentrations of o,p'DDD, o,p'DDA and o,p'DDE as predictors
of tumor response to mitotane in adrenocortical carcinoma: results
of a retrospective ENS@T multicenter study. J Clin Endocrinol
Metab. 96:1844–1851. 2011.
|
|
4
|
Allolio B and Fassnacht M: Clinical
review: Adrenocortical carcinoma: clinical update. J Clin
Endocrinol Metab. 91:2027–2037. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Libe R, Fratticci A and Bertherat J:
Adrenocortical cancer: pathophysiology and clinical management.
Endocr Relat Cancer. 14:13–28. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kirschner LS: Emerging treatment
strategies for adrenocortical carcinoma: a new hope. J Clin
Endocrinol Metab. 91:14–21. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zini L, Porpiglia F and Fassnacht M:
Contemporary management of adrenocortical carcinoma. Eur Urol.
60:1055–1065. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haak HR, Hermans J, van de Velde CJ,
Lentjes EG, Goslings BM, Fleuren GJ and Krans HM: Optimal treatment
of adrenocortical carcinoma with mitotane: results in a consecutive
series of 96 patients. Br J Cancer. 69:947–951. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gicquel C, Bertagna X, Gaston V, Coste J,
Louvel A, Baudin E, Bertherat J, Chapuis Y, Duclos JM, Schlumberger
M, Plouin PF, Luton JP and Le Bouc Y: Molecular markers and
long-term recurrences in a large cohort of patients with sporadic
adrenocortical tumors. Cancer Res. 61:6762–6767. 2001.PubMed/NCBI
|
|
10
|
Ragazzon B, Libe R, Gaujoux S, Assie G,
Fratticci A, Launay P, Clauser E, Bertagna X, Tissier F, de Reynies
A and Bertherat J: Transcriptome analysis reveals that p53 and
{beta}-catenin alterations occur in a group of aggressive
adrenocortical cancers. Cancer Res. 70:8276–8281. 2010.
|
|
11
|
Asp V, Ulleras E, Lindstrom V, Bergstrom
U, Oskarsson A and Brandt I: Biphasic hormonal responses to the
adrenocorticolytic DDT metabolite 3-methylsulfonyl-DDE in human
cells. Toxicol Appl Pharmacol. 242:281–289. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin CW, Chang YH and Pu HF: Mitotane
exhibits dual effects on steroidogenic enzymes gene transcription
under basal and cAMP-stimulating microenvironments in NCI-H295
cells. Toxicology. 298:14–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gilep AA, Sushko TA and Usanov SA: At the
crossroads of steroid hormone biosynthesis: the role, substrate
specificity and evolutionary development of CYP17. Biochim Biophys
Acta. 1814:200–209. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chung BC, Picado-Leonard J, Haniu M,
Bienkowski M, Hall PF, Shively JE and Miller WL: Cytochrome P450c17
(steroid 17 alpha-hydroxylase/17,20 lyase): cloning of human
adrenal and testis cDNAs indicates the same gene is expressed in
both tissues. Proc Natl Acad Sci USA. 84:407–411. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miller WL: Minireview: regulation of
steroidogenesis by electron transfer. Endocrinology. 146:2544–2550.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Staels B, Hum DW and Miller WL: Regulation
of steroidogenesis in NCI-H295 cells: a cellular model of the human
fetal adrenal. Mol Endocrinol. 7:423–433. 1993.PubMed/NCBI
|
|
17
|
Ahlman H, Khorram-Manesh A, Jansson S,
Wangberg B, Nilsson O, Jacobsson CE and Lindstedt S: Cytotoxic
treatment of adrenocortical carcinoma. World J Surg. 25:927–933.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pushkarev VM, Tronko ND, Kostyuchenko NN
and Mikosha AS: Effect of o,p'-DDD and Li+ on apoptotic
DNA fragmentation in conventionally normal and tumour tissues of
human adrenal cortex. Ukr Biokhim Zh. 79:44–49. 2007.
|
|
19
|
de Fraipont F, El Atifi M, Cherradi N, Le
Moigne G, Defaye G, Houlgatte R, Bertherat J, Bertagna X, Plouin
PF, Baudin E, Berger F, Gicquel C, Chabre O and Feige JJ: Gene
expression profiling of human adrenocortical tumors using
complementary deoxyribonucleic acid microarrays identifies several
candidate genes as markers of malignancy. J Clin Endocrinol Metab.
90:1819–1829. 2005.
|
|
20
|
Dohna M, Reincke M, Mincheva A, Allolio B,
Solinas-Toldo S and Lichter P: Adrenocortical carcinoma is
characterized by a high frequency of chromosomal gains and
high-level amplifications. Genes Chromosomes Cancer. 28:145–152.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kjellman M, Kallioniemi OP, Karhu R, Hoog
A, Farnebo LO, Auer G, Larsson C and Backdahl M: Genetic
aberrations in adrenocortical tumors detected using comparative
genomic hybridization correlate with tumor size and malignancy.
Cancer Res. 56:4219–4223. 1996.
|
|
22
|
Stephan EA, Chung TH, Grant CS, Kim S, Von
Hoff DD, Trent JM and Demeure MJ: Adrenocortical carcinoma survival
rates correlated to genomic copy number variants. Mol Cancer Ther.
7:425–431. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moore CC, Hum DW and Miller WL:
Identification of positive and negative placenta-specific basal
elements and a cyclic adenosine 3′,5′-monophosphate response
element in the human gene for P450scc. Mol Endocrinol. 6:2045–2058.
1992.PubMed/NCBI
|
|
24
|
Sewer MB, Nguyen VQ, Huang CJ, Tucker PW,
Kagawa N and Waterman MR: Transcriptional activation of human CYP17
in H295R adrenocortical cells depends on complex formation among
p54(nrb)/NonO, protein-associated splicing factor and SF-1, a
complex that also participates in repression of transcription.
Endocrinology. 143:1280–1290. 2002. View Article : Google Scholar
|
|
25
|
Asp V, Lindstrom V, Olsson JA, Bergstrom U
and Brandt I: Cytotoxicity and decreased corticosterone production
in adrenocortical Y-1 cells by 3-methylsulfonyl-DDE and
structurally related molecules. Arch Toxicol. 83:389–396. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi YQ, Li HW, Wang YP, Liu CJ and Yang
KD: p,p'-DDE induces apoptosis and mRNA expression of
apoptosis-associated genes in testes of pubertal rats. Environ
Toxicol. Mar 7–2011.(Epub ahead of print).
|
|
27
|
Burow ME, Tang Y, Collins-Burow BM,
Krajewski S, Reed JC, McLachlan JA and Beckman BS: Effects of
environmental estrogens on tumor necrosis factor alpha-mediated
apoptosis in MCF-7 cells. Carcinogenesis. 20:2057–2061. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boatright KM and Salvesen GS: Mechanisms
of caspase activation. Curr Opin Cell Biol. 15:725–731. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lakhani SA, Masud A, Kuida K, Porter GA
Jr, Booth CJ, Mehal WZ, Inayat I and Flavell RA: Caspases 3 and 7:
key mediators of mitochondrial events of apoptosis. Science.
311:847–851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alenzi FQ, Lotfy M and Wyse R: Swords of
cell death: caspase activation and regulation. Asian Pac J Cancer
Prev. 11:271–280. 2010.PubMed/NCBI
|
|
31
|
Fuentes-Prior P and Salvesen GS: The
protein structures that shape caspase activity, specificity,
activation and inhibition. Biochem J. 384:201–232. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hogel H, Rantanen K, Jokilehto T, Grenman
R and Jaakkola PM: Prolyl hydroxylase PHD3 enhances the hypoxic
survival and G1 to S transition of carcinoma cells. PLoS One.
6:e271122011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wilhelm S, Wagner H and Hacker G:
Activation of caspase-3-like enzymes in non-apoptotic T cells. Eur
J Immunol. 28:891–900. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Feige JJ, Cochet C and Chambaz EM: Type
beta transforming growth factor is a potent modulator of
differentiated adrenocortical cell functions. Biochem Biophys Res
Commun. 139:693–700. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Riopel L, Branchaud CL, Goodyer CG, Adkar
V and Lefebvre Y: Growth-inhibitory effect of TGF-B on human fetal
adrenal cells in primary monolayer culture. J Cell Physiol.
140:233–238. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ragazzon B, Cazabat L, Rizk-Rabin M, Assie
G, Groussin L, Fierrard H, Perlemoine K, Martinez A and Bertherat
J: Inactivation of the Carney complex gene 1 (protein kinase A
regulatory subunit 1A) inhibits SMAD3 expression and TGF
beta-stimulated apoptosis in adrenocortical cells. Cancer Res.
69:7278–7284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Khaidakov M, Szwedo J, Mitra S and Mehta
JL: Angiostatic effects of aspirin in hypoxia-reoxygenation are
linked to modulation of TGFbeta1 signaling. J Cardiovasc Pharmacol
Ther. 16:105–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nieminen AI, Partanen JI and Klefstrom J:
c-Myc blazing a trail of death: coupling of the mitochondrial and
death receptor apoptosis pathways by c-Myc. Cell Cycle.
6:2464–2472. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Prendergast GC: Mechanisms of apoptosis by
c-Myc. Oncogene. 18:2967–2987. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gartel AL and Tyner AL: The role of the
cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer
Ther. 1:639–649. 2002.PubMed/NCBI
|
|
41
|
Shin H, Lee YS and Lee YC: Sodium
butyrate-induced DAPK-mediated apoptosis in human gastric cancer
cells. Oncol Rep. 27:1111–1115. 2012.PubMed/NCBI
|
|
42
|
Suzuki A, Kawano H, Hayashida M, Hayasaki
Y, Tsutomi Y and Akahane K: Procaspase 3/p21 complex formation to
resist fas-mediated cell death is initiated as a result of the
phosphorylation of p21 by protein kinase A. Cell Death Differ.
7:721–728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tait SW and Green DR: Mitochondria and
cell death: outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen CW, Hurd C, Vorojeikina DP, Arnold SF
and Notides AC: Transcriptional activation of the human estrogen
receptor by DDT isomers and metabolites in yeast and MCF-7 cells.
Biochem Pharmacol. 53:1161–1172. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gell JS, Oh J, Rainey WE and Carr BR:
Effect of estradiol on DHEAS production in the human adrenocortical
cell line, H295R. J Soc Gynecol Investig. 5:144–148. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Vanttinen T, Liu J, Kuulasmaa T, Kivinen P
and Voutilainen R: Expression of activin/inhibin signaling
components in the human adrenal gland and the effects of activins
and inhibins on adrenocortical steroidogenesis and apoptosis. J
Endocrinol. 178:479–489. 2003. View Article : Google Scholar : PubMed/NCBI
|