Introduction
The acute administration of ethanol to intestinal
epithelial cells causes increased intestinal permeability and the
translocation of endotoxin, which is a component of the outer wall
of Gram-negative bacteria and is normally excluded by the
intestinal barrier. Despite several studies, the pathogenesis of
the effects of ethanol on the small intestine is not yet clear
(1–3). One study indicated that structural
and functional abnormalities caused by ethanol are associated with
oxidative stress and this was suggested as the mechanism by which
ethanol increases intestinal permeability (4). This model has recently been
demonstrated in lung cells (5).
Increases in reactive oxygen species (ROS) by oxidative stress
increase lipid, protein and DNA peroxidation. ROS from xenobiotics,
including ethanol, cause oxidative DNA damage through single-strand
breaks (6). According to Bradford
et al (7), when ethanol is
administered to the liver of rats, the expression of DNA repair
enzymes increases and may be used as a sensitive DNA damage marker.
Interest in the small intestine has been on the increase due to the
development of capsule endoscopy and enteroscopy (8). However, only a few studies have been
conducted on DNA damage and repair associated with oxidative stress
caused by ethanol in the small intestine.
Base excision repair (BER) is the main DNA repair
mechanism, which maintains the stability of the genome in response
to DNA damage caused by reactive chemicals that are constantly
created (9). A variety of
repair-related enzymes and proteins, including DNA polymerase β,
apurinic/apyrimidinic endonuclease/redox factor-1 (APE/Ref-1),
proliferating cell nuclear antigen (PCNA) and growth arrest and DNA
damage 45α (GADD45α) are used in BER. However, DNA damage and
repair-related enzymes in intestinal cells which act against
stresses including ethanol have not been fully investigated.
Heat shock proteins (Hsps) are cell proteins that
remove or repair denatured proteins and prevent protein
aggregation. Hsps are involved in recovery and protection from cell
damage. Hsps are associated with DNA repair enzymes and related
proteins during cellular oxidative damage (10,11).
Therefore, the aim of our study was to investigate
cytotoxicity, DNA damage and the expression of DNA repair-related
molecules, antioxidant proteins and Hsps in intestinal cells
exposed to ethanol for a short time.
Materials and methods
Chemicals
Urea, thiourea,
3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate,
dithiothreitol, acrylamide, NN′-methylene-bisacrylamide, tris and
sodium dodecyl sulfate (SDS) were purchased from Sigma Chemical
(St. Louis, MO, USA). Ethanol was purchased from Merck (Merck Co.,
Darmstadt, Germany).
Cell culture
Caco-2 cells, a human colorectal adenocarcinoma cell
line, were obtained from American Type Culture Collection
(ATCC-HTB-37TM; Manassas, VA, USA). These cells form
polarized monolayers that morphologically and functionally resemble
adult human small intestinal mucosal cells subsequent to reaching
confluence. Cells were maintained in MEM containing 20% fetal
bovine serum albumin (ATCC), penicillin (100 U/ml) and streptomycin
(100 μg/ml) at 37˚C in a 5% CO2 in air atmosphere. Cells
cultured for 1 h with 1, 2, 3, 4, 5, 6, 7 and 8% ethanol were used
for the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium
bromide (MTT) and comet assays, as well as in western blot
analysis.
MTT assay
Caco-2 cells (1×104) were incubated with
different concentrations (1, 2, 3, 4, 5, 6, 7 and 8%) of ethanol in
96-well plates for 1 h and cell viability was determined using the
MTT assay (Calbiochem, San Diego, CA, USA) (12). Briefly, 20 μl of 5 mg/ml MTT in
phosphate-buffered saline (PBS; Gibco BRL, Paisley, Scotland, UK)
was added to each well and incubated for 3 h at 37˚C. The media
were then removed and formazan crystals in the cells were dissolved
in the presence of 200 μl lysis buffer (5% w/v of SDS in 0.01 N
HCl). The plates were read at a 590 nm wavelength using an ELISA
reader (Molecular Devices Co., Sunnyvale, CA, USA). The percentage
of cell proliferation and cytotoxicity was determined by comparing
the optical densities of the cells treated with different
concentrations of ethanol with that of the control. Each experiment
was repeated seven times.
Comet assay
DNA damage was determined using the comet assay.
This assay was performed according to Singh et al with minor
modifications (13). Briefly,
normal and low melting point agarose (NMA and LMA, respectively;
Ameresco, Solon, OH, USA) was added to fully frosted slides that
were precoated with 50 μl 1% NMA for firm attachment and the slides
were then allowed to solidify with cover slips in the refrigerator
for 5 min. Following solidification of the gel, the cover slips
were removed and 50 μl cells mixed with 50 μl of 1% LMA were added.
The cover slips were added on to the layer and the slides were
allowed to solidify in the refrigerator for 5 min. Following the
removal of the cover slips, 100 μl of 0.5% LMA was added as a third
layer and the slides were again placed with cover slips in the
refrigerator for 5 min. The slides were submersed in a lysing
solution (2.5 M NaCl, 100 mM EDTA-2Na, 10 mM Tris-HCl, pH 10; 1%
Triton X-100 and 10% DMSO, pH 10 added fresh) for 1 h. The slides
were then placed in an unwinding buffer (1 mM EDTA and 300 mM NaOH,
pH 13) for 20 min and electrophoresis was performed using the same
solution for 20 min at 25 V and 300 mA (0.8 v/cm). Following
electrophoresis, the slides were neutralized by washing three times
with neutralization buffer (400 mM Tris-HCl, pH 7.4) for 5 min each
and were then stained with 50 μl of 10 μg/ml ethidium bromide. The
slides were examined using a Komet 4.0 image analysis system
(Kinetic Imaging, Liverpool, UK) fitted with an Olympus BX50
fluorescence microscope (Olympus, Tokyo, Japan) and equipped with a
515–560 nm excitation filter and a 590 nm barrier filter. Two
slides were prepared for each treatment group and 50 randomly
selected cells (total 100 cells) were scored manually. The
parameter Olive tail moment [=(Tail.mean-Head.mean) × Tail%DNA/100]
was automatically calculated using the Komet 4.0 image analysis
system, which was used for global comet description. Each
experiment was repeated three times.
Western blot assay
Caco-2 cells were solubilized in a lysis buffer (pH
7.4) on ice using a homogenizer. The lysates were then clarified by
centrifugation at 12,000 rpm for 15 min at 4˚C and the protein
concentration of the total lysate was determined using a Bradford
protein assay (Bio-Rad Laboratories, Richmond, CA, USA). An equal
amount of protein per lane was separated by electrophoresis on 8
and 15% SDS-polyacrylamide gels and then transferred to
polyvinylidene difluoride membranes (Millipore Corporation,
Bedford, MA, USA) at 400 mA for 160 min using a transfer buffer (pH
8.3). The membranes were blocked with blocking buffer (PBS
containing 5% skimmed milk) for 1 h at room temperature, followed
by incubation with primary antibodies overnight at 4˚C. Subsequent
to the membranes being washed three times with PBS-T for 20 min,
they were further incubated with horseradish peroxidase-conjugated
secondary antibodies [anti-rabbit IgG and anti-mouse IgG (1:2,000;
Santa Cruz Biotechnology, Santa Cruz, CA, USA)] for 1 h at room
temperature and washed three times for 20 min with PBS-T. Following
extensive washing, the immune complexes were then detected using
the enhanced chemiluminescence (ECL) and ECL Plus systems (Amersham
Pharmacia Biotech, Piscataway, NJ, USA). Primary antibodies against
DNA polymerase β (1:1,000; Abcam, Cambridge, MA, USA), APE/Ref-1
(1:4,000; Santa Cruz Biotechnology), PCNA (1:100; Santa Cruz
Biotechnology), GADD45α (1:500; Santa Cruz Biotechnology),
glutathione peroxidase-1 (GPx-1; 1:1,000; Abcam), peroxiredoxin-1
(PRX-1; 1:1,000; Santa Cruz Biotechnology), superoxide dismutase-2
(SOD-2; 1:10,000; Abcam), Hsp10 (1:20,000; Abcam), Hsp27 (1:1,000;
Cell Signaling Technology, Inc., Danvers/Beverly, MA, USA), Hsp60
(1:1,000; Santa Cruz Biotechnology), Hsp70 (1:1,000; Biosciences,
San Diego, CA, USA), heat shock cognate (Hsc)70 (1:2,000; Santa
Cruz Biotechnology) and Hsp90 (1:4,000; BD Transduction
Laboratories) were applied at the optimal concentrations. Bands
were visualized by ECL and scanned using a flat-bed scanner. The
digitalized images were analyzed using Scion image analysis
software (Scion Co., Frederick, MD, USA).
Statistical analysis
Variables were presented as the mean ± SD and the
statistical analysis was performed using an analysis of variance
followed by Dunnett's test. P<0.05 was considered to indicate
statistically significant results. Data were analyzed using
statistical software (SPSS for Windows version 12.0; SPSS Inc.,
Chicago, IL, USA).
Results
Cytotoxicity
The exposure of Caco-2 cells to 1–8% ethanol for 1 h
resulted in a dose-dependent decrease in viability as measured by
the MTT assay. The effect was statistically significant at ethanol
concentrations ≥7% (P<0.05; Fig.
1).
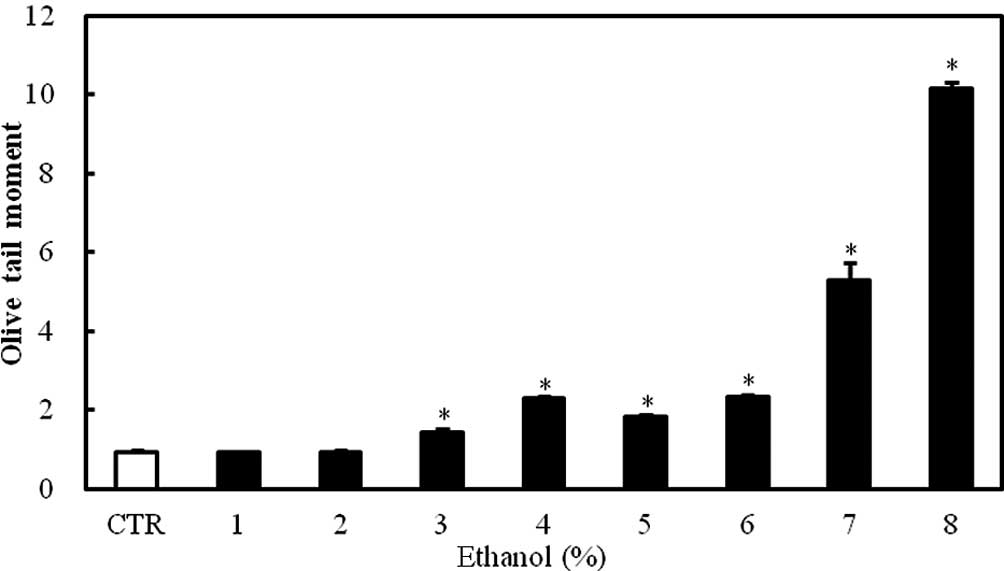
DNA damage
The exposure of Caco-2 cells to 1–8% ethanol for 1 h
caused a dose-dependent increase in the Olive tail moment, which is
a DNA damage parameter in the comet assay. The effect was
statistically significant at ethanol concentrations ≥3% (P<0.05;
Fig. 2).
Effect of ethanol on DNA repair-related
molecules
Western blot analyses were performed to evaluate the
expression of the BER pathway DNA repair-related molecules,
including DNA polymerase β, PCNA, APE/Ref-1 and GADD45α, in Caco-2
cells exposed to graded ethanol concentrations (1, 2, 4 and 8%) for
1 h.
The relative intensities revealed that PCNA and
APE/Ref-1 expression increased significantly in Caco-2 cells
exposed to 1% ethanol, the level of DNA polymerase β expression
increased in 4% ethanol and GADD45α expression did not increase
significantly in low concentrations of ethanol. The expression of
PCNA, APE/Ref-1, DNA polymerase β and GADD45α all decreased at 8%
ethanol (Fig. 3).
Effect of ethanol on antioxidant
enzymes
Western blot analyses were performed to evaluate the
expression of the antioxidant enzymes GPx-1, PRX-1 and SOD-2 in
Caco-2 cells exposed to graded ethanol concentrations (1, 2, 4 and
8%) for 1 h.
The relative intensities revealed that GPx-1, PRX-1
and SOD-2 expression increased significantly in Caco-2 cells
exposed to 1% ethanol; however, their expression decreased with the
8% ethanol exposure (Fig. 4).
Effect of ethanol on Hsps
Western blot analyses revealed the level of Hsp
expression in Caco-2 cells exposed to graded ethanol concentrations
(1, 2, 4 and 8%) for 1 h. The relative intensities of Hsp10, Hsp27
and Hsp70 expression increased significantly at 1% ethanol. Hsp10
decreased significantly at 8% ethanol. Hsp27 and Hsp70 also
decreased gradually with further increases in ethanol concentration
(Fig. 5).
 | Figure 5Effect of ethanol on Hsps in Caco-2
cells. (A) Western blot analysis of Hsp10, Hsp27, Hsp60, Hsc70,
Hsp70 and Hsp90. (B) Quantitation of the relative expression
intensity of Hsp10, Hsp27, Hsp60, Hsc70, Hsp70 and Hsp90. Values
are mean ± SD. *P<0.05 versus control (CTR). Hsp,
heat shock protein; Hsc, heat shock cognate. |
However, Hsp60 expression increased significantly at
a concentration of 4% ethanol. Hsc70 and Hsp90 expression increased
with 2% ethanol exposure. Hsc70 expression decreased significantly
with 8% ethanol exposure (Fig.
4).
Discussion
This study examined the cytotoxicity, DNA damage and
the expression of DNA repair-related molecules, antioxidants and
Hsps in intestinal epithelial cells exposed to low concentrations
of ethanol for 1 h.
Acute and chronic exposure of the small intestine to
ethanol causes various structural and functional abnormalities. The
acute administration of ethanol increases intestinal permeability
within 30 min and causes the transfer of endotoxin into the blood,
leading to endotoxemia. Intermittent endotoxemia then stimulates
Kupffer cells in the liver and facilitates the formation of ROS and
inflammatory mediators, resulting in liver damage.
In our study, 3 and 7% concentrations of ethanol
induced statistically significant DNA damage and cytotoxicity,
respectively. Therefore, low ethanol concentrations (<10%) may
cause intestinal epithelial cell death, as reported in previous
studies (14,15). Additionally, genotoxicity may be
associated with cytotoxicity. Based on these results, the ethanol
concentrations required to observe the expression of proteins and
enzymes involved in DNA repair in intestinal epithelial cells were
determined. Accordingly, Caco-2 cells were exposed to 1, 2, 4 and
8% graded ethanol concentrations for 1 h.
The mechanism of BER is as follows (13): DNA glycosylase removes damaged DNA
bases by cutting the N-glycosyl bond. The apurinic or apyrimidinic
site lacking the DNA base is known as the abasic or the AP site. AP
endonuclease (another name for APE/Ref-1) cleaves the
phosphodiester backbone near the AP site. DNA polymerase then
removes the damaged part of the strand and synthesizes a new
strand. Unlike short-patch repair, in which DNA polymerase β
inserts a single nucleotide, if the AP site is oxidized or reduced,
it is resistant to DNA polymerase β and long-patch repair occurs.
During long-patch repair, DNA polymerases δ and ɛ act with PCNA,
which is a repair-related protein. APE/Ref-1 is a significant
indicator of DNA repair enzymes and an essential element in cancer
research. GADD45α is the transcriptional target of p53 and delays
carcinogenesis and decreases mutation frequency. A recent study
suggested that GADD45α improves the BER response by binding to DNA
and influencing the interaction between cellular APE/Ref-1 and PCNA
(16). More research concerning
the role of these proteins in DNA damage-repair mechanisms is
needed.
In this study, the level of DNA repair-related
molecules increased in intestinal epithelial cells exposed to low
concentrations of ethanol for a short time. First, PCNA and
APE/Ref-1 increased in the 1% ethanol-exposed cells. DNA polymerase
β increased at higher concentrations than PCNA and APE/Ref-1.
GADD45α did not show a significant increase. Therefore, PCNA and
APE/Ref-1 may be useful in evaluating the level of DNA damage in
intestinal epithelial cells. In a previous study, which evaluated
DNA damage in human lymphocytes exposed to hydrogen peroxide
(H2O2) and methyl methane sulfonate (MMS) for
a short time, GADD45α was more sensitive to DNA damage than other
repair-related molecules, including DNA polymerase β, PCNA and
APE/Ref-1 (17). However,
different expression patterns of repair-related molecules in Caco-2
cells compared with lymphocytes were observed in this study.
Therefore, although the toxicants used were different, the response
to genotoxic compounds may vary according to cell type.
All antioxidant enzymes, including GPx-1, PRX-1 and
SOD-2, increased in 1% ethanol-exposed cells, indicating that
oxidative stress is involved in the intestinal cell damage caused
by ethanol. Oxidative stress may induce the synthesis of Hsps. Hsps
function as molecular chaperones by facilitating protein synthesis,
folding, transport, translocation and processing. Hsps are
therefore essential for cell survival (5). Hsps are classified into subfamilies
depending on their molecular weight, including Hsp60, Hsp70, Hsp90
and the small Hsp family (18).
Among the Hsps, the expression of Hsp10, Hsp27 and
Hsp70 increased significantly at 1% ethanol. Hsc70 and Hsp90 also
had significant changes in expression at higher concentrations than
those of Hsp10, Hsp27 and Hsp70. Hsp60 also had significant changes
in the levels of expression. Different Hsps are induced in
different organs. In intestinal epithelial cells, the Hsp70 family
and Hsp25 are induced (19,20).
Hsp60 has no protective role in the intestine and Hsp10 is a
molecular chaperone that functions in protein stabilization and
folding with Hsp60 (21). The
overexpression of Hsp10 has been reported at a higher rate than
Hsp70 in cancer cells, including colorectal cancer (22). It is speculated that Hsp10 affects
cell life and death unlike Hsp60, which does not affect cell life
and death. In other words, Hsp10 is considered an active factor in
the cell signaling pathway and affects the cell cycle,
nucleocytoplasmic transport and metabolism (23).
DNA repair-related molecules and Hsps had similar
expression patterns based on the ethanol concentration. Their
expression intially increased with the increasing ethanol
concentration. Expression of most of these molecules significantly
changed at an ethanol concentration <3%, at which point
significant DNA damage occurred. However, the expression of all the
repair-related molecules decreased at an 8% ethanol concentration,
suggesting that intestinal epithelial cells lose their repair
capacity due to cell death, which occurred at a 7% ethanol
concentration in the MTT assay.
It could be inferred that Hsps are associated with
DNA repair. Mendez et al (11) revealed that Hsp70 stimulates the
BER enzyme, DNA polymerase β. Kenny et al (10) stated that Hsp70 binds to human AP
endonuclease and stimulates endonuclease activity at abasic sites.
These studies suggest that a single-strand DNA binding protein may
be needed for BER and that Hsps may play a role as BER accessory
proteins. If DNA repair-related molecules could be measured
according to the induction or suppression of Hsps, they may be
useful for clarifying the association between DNA repair and
Hsps.
The limitation of our study was the use of a tumor
cell line that was not suited to showing physiological phenomena.
Although Caco-2 cells originated from human colorectal
adenocarcinoma cells, they have similar characteristics to small
intestinal epithelial cells. In addition, normal intestinal cell
lines, including FHs74Int, are difficult to culture. Therefore,
Caco-2 cells were used to investigate the heat shock response and
gene expression in this study.
In conclusion, the results revealed that acute and
low (<10%) concentrations of ethanol induced DNA damage and
cytotoxicity in human intestinal epithelial Caco-2 cells. In
particular, PCNA, APE/Ref-1, Hsp10, Hsp27 and Hsp70 were more
sensitive to ethanol than the other DNA repair molecules and Hsps.
These proteins may be useful in evaluating the genotoxicity of
toxicants and the DNA repair and cytoprotective effects of drugs in
intestinal cells under stress.
References
|
1
|
C BodeJC BodeEffect of alcohol consumption
on the gutBest Pract Res Clin
Gastroenterol17575592200310.1016/S1521-6918(03)00034-912828956
|
|
2
|
L BujandaThe effects of alcohol
consumption upon the gastrointestinal tractAm J
Gastroenterol9533743382200010.1111/j.1572-0241.2000.03347.x11151864
|
|
3
|
K NakagawaJ AdachiMCY WongY UenoProtective
effect of daidzein against acute ethanol-induced lipid peroxidation
in rat jejunumKobe J Med Sci52141149200617006054
|
|
4
|
A FarhadiA KeshavarzianZ RanjbaranJZ
FieldsA BananThe role of protein kinase C isoforms in modulating
injury and repair of the intestinal barrierJ Pharmacol Exp
Ther31617200610.1124/jpet.105.08544916002462
|
|
5
|
LA BrownFL HarrisXD PingTW GauthierChronic
ethanol ingestion and the risk of acute lung injury: a role for
glutathione
availability?Alcohol33191197200410.1016/j.alcohol.2004.08.00215596087
|
|
6
|
U Mutlu-TurkogluS Dogru-AbbasogluG
Aykac-TokerH MirsalM BeyazyurekM UysalIncreased lipid and protein
oxidation and DNA damage in patients with chronic alcoholismJ Lab
Clin Med136287291200010.1067/mlc.2000.10909711039849
|
|
7
|
BU BradfordH KonoF IsayamaCytochrome P450
CYP2E1, but not nicotinamide adenine dinucleotide phosphate
oxidase, is required for ethanol-induced oxidative DNA damage in
rodent liverHepatology41336344200510.1002/hep.20532
|
|
8
|
CA TennysonCE SemradAdvances in small
bowel imagingCurr Gastroenterol
Rep13408417201110.1007/s11894-011-0221-921845375
|
|
9
|
M ChristmannMT TomicicWP RoosB
KainaMechanisms of human DNA repair: an
updateToxicology193334200310.1016/S0300-483X(03)00287-714599765
|
|
10
|
MK KennyF MendezM SandigurskyHeat shock
protein 70 binds to human apurinic/apyrimidinic endonuclease and
stimulates endonuclease activity at abasic sitesJ Biol
Chem27695329536200110.1074/jbc.M00929720011133992
|
|
11
|
F MendezE KozinR BasesHeat shock protein
70 stimulation of the deoxyribonucleic acid base excision repair
enzyme polymerase betaCell Stress
Chaperones8153161200310.1379/1466-1268(2003)008%3C0153:HSPSOT%3E2.0.CO;214627201
|
|
12
|
T MosmannRapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assaysJ Immunol
Methods655563198310.1016/0022-1759(83)90303-46606682
|
|
13
|
NP SinghMT McCoyRR TiceEL SchneiderA
simple technique for quantitation of low levels of DNA damage in
individual cellsExp Cell
Res175184191198810.1016/0014-4827(88)90265-0
|
|
14
|
K AsaiWA BuurmanCP ReutelingspergerB
SchutteM KaminishiLow concentrations of ethanol induce apoptosis in
human intestinal cellsScand J
Gastroenterol3811541161200310.1080/0036552031000625214686719
|
|
15
|
TY MaD NguyenV BuiH NguyenN HoaEthanol
modulation of intestinal epithelial tight junction barrierAm J
Physiol276G965G974199910198341
|
|
16
|
HJ JungEH KimJY MunBase excision DNA
repair defect in Gadd45a-deficient
cellsOncogene2675177525200710.1038/sj.onc.121055717599061
|
|
17
|
EK ChoSY ParkDG SulComparison of DNA
damage and the expression of repair related molecules, including
DNA polymerase β, APE/ref-1, PCNA, and GADD45, in human T and B
lymphocytes exposed to hydrogen peroxide and methyl
methanesulfonateNew Research on DNA DamageH KimuraA SuzukaNova
Science Publishers, IncHauppauge, NY305318200822246134
|
|
18
|
M TominagaM OhtaS KaiK IwakiK ShibataS
KitanoIncreased heat-shock protein 90 expression contributes to
impaired adaptive cytoprotection in the gastric mucosa of portal
hypertensive ratsJ Gastroenterol
Hepatol2411361141200910.1111/j.1440-1746.2008.05763.x19383083
|
|
19
|
S OtaniM OtakaM JinEffect of preinduction
of heat shock proteins on acetic acid-induced colitis in ratsDig
Dis Sci42833846199710.1023/A:10188326182759125658
|
|
20
|
MJ RopeleskiJ TangMM Walsh-ReitzMW MuschEB
ChangInterleukin-11-induced heat shock protein 25 confers
intestinal epithelial-specific cytoprotection from oxidant
stressGastroenterology12413581368200310.1016/S0016-5085(03)00282-812730876
|
|
21
|
A IwabuchiM OtakaS OtaniSpecific
preinduction of 60-kDa heat shock protein (chaperonin homolog) by
TRH does not protect colonic mucosa against acetic acid-induced
lesion in ratsDig Dis
Sci4514801489200010.1023/A:100559711302410961734
|
|
22
|
C JunhuiC LimingW ShaobinH JiexiongQ
QianchengX LiyanClinical significance of heat shock protein 10 in
large bowel carcinomaChinese-German J Clin
Oncol6334338200710.1007/s10330-007-0032-5
|
|
23
|
V StribinskisHC HeymanSR EllisMC SteffenNC
MartinRpm2p, a component of yeast mitochondrial RNase P, acts as a
transcriptional activator in the nucleusMol Cell
Biol2565466558200510.1128/MCB.25.15.6546-6558.200516024791
|