Introduction
Members of the transforming growth factor (TGF)-β
family, including TGF-β, activin, nodal and bone morphogenetic
proteins (BMPs), are multifunctional cytokines that regulate a wide
range of cellular responses, such as cellular proliferation,
adhesion and differentiation, haematopoiesis, inflammation, wound
repair and skeletal development (1). BMPs were identified based on their
ability to promote ectopic cartilage and bone formation (2). BMPs function through conserved type I
and type II transmembrane receptors and Smad-dependent and
-independent pathways, to regulate a range of biological processes
in a highly context-dependent manner (1,3–5).
Disruption of these pathways can lead to various diseases including
cancer (6).
Estrogenic hormones regulate multiple activities,
including cell proliferation and differentiation, in different
types of cells. It is widely accepted that the hormone-occupied
estrogen receptor (ER) functions as a versatile transcription
factor to either activate or repress gene expressions (7). These effects of estrogen on
transcriptional regulation involve both the direct interaction of
ER with DNA encoded estrogen response elements (EREs) and the
indirect tethering of ER to DNA through protein-protein
interactions (8,9). In addition to these nuclear events,
estrogen is capable of evoking rapid, membrane-initiated signaling
events, such as the release of calcium, secretion of prolactin,
generation of nitric oxide, regulation of PI3K/Akt and the
activation of the MAPK pathway (10–12).
Estrogen can influence these effects in a variety of cell types
(13), with the exact response
dependent on the nature of the target cell.
The ER signaling pathway plays a pivotal role in the
development of different types of breast cancer (14). Two types of ERs have been
identified, ERα and ERβ, both of which have many mRNA splice
variants (15). For example, there
are 3 types of ERα isoforms identified: 66-KD, 46-KD and 36-KD
(16). Usually, if a cell line
expresses ERα-66 (e.g., MCF-7 cells) it is considered ERα-positive;
if a cell line does not express ERα-66 (e.g., MDA-MB-231 cells) it
is termed ERα-negative.
Endocrine therapy is effective in approximately
one-third of all breast cancers, although up to 80% of these
express both estrogen and progesterone receptors (17). Unfortunately, most breast cancer
cells acquire resistance due to the use of steroid hormones in the
process of endocrine treatment for controlling the growth of cancer
cells (18). In order to
understand the effects of ERs on the development of breast cancer,
we wished to explore the correlation between the ER signaling
pathway and other pathways, such as the Wnt/wingless, receptor
tyrosine kinase, JAK/STAT and the BMP signaling pathways, which are
included among the conserved pathways that control the fate of
cells (19,20).
In this study, we report for the first time that
BMP2 induces the expression of ERα-36, but not ERα-66, in
MDA-MB-231 and MCF-7 breast cancer cell lines. The results from our
study indicate that BMP2 alters the expression profile of ERα and
thus, has the potential to alter the response of breast cancer
cells to endocrine therapy.
Materials and methods
Cell lines and antibodies
The human breast cancer cell lines, MDA-MB-231
(ER-negative) and MCF-7 (ER-positive), were obtained from the
American Type Culture Collection (ATCC). All cells were passaged
for a period of <6 months subsequent to resuscitation, and
cultured using the protocol provided by ATCC. The sera and media
were purchased from Invitrogen and ATCC, whereas anti-β-actin,
anti-ERα-66 and anti-ERβ antibodies were from Cell Signaling
Technology. HRP-goat anti-rabbit conjugate and HRP-goat anti-mouse
conjugate were purchased from Santa Cruz Biotechnology,.
Quantitative RT-PCR
MCF-7 and MDA-MB-231 breast cancer cells were plated
in 10-cm dishes, and subsequently treated with BMP2 (20 ng/ml) for
0, 24 and 48 h. Total RNA was extracted using TRIzol reagent, and
cDNA was prepared using SuperScript II Reverse Transcriptase
(Invitrogen). Quantitative RT-PCR was performed using the IQ
SYBR-Green Mix in an iCycler PCR machine (Bio-Rad), using 1 μl of
cDNA in triplicate. Primers used are included in Table I.
 | Table IPrimers used for real-time PCR. |
Table I
Primers used for real-time PCR.
| Genes | Primer sequences (F,
forward; R, reverse) |
|---|
| ERα | F:
AAGTATTCAAGGACATAACG | R:
TATCCCACCTTTCATCAT |
| ERα-66 | F:
GGTGCCCTACTACCTGGAGA | R:
TCTGAATTTGGCCTGTAGAATG |
| ERα-36 | F:
GACAGGAACCAGGGAAAA | R:
TCTACATGTGAGATACCAGA |
| ERβ | F:
TCATGAATTACAGCATTCCC | R:
ATGAAGTGAGCATCCCTCTT |
| BMPR1b | F:
AATGCCACCATTGTCCA | R:
CTAGGCAACCAGAAGTGACCACAG |
| BMPR1a | F:
AGTGTCTCCAGTCAAGCTCTGGGTA | R:
CCATCTCTGCTGCTGCGCTCATTTA |
| BMPR2 | F:
TGCAGATGGACGCATGGAA | R:
CGGCAAGAGCTTACCCAGTCA |
| p21 | F:
TGAGCCGCGACTGTGATG | R:
GTCTCGGTGACAAAGTCGAAGTT |
Specific antibody for ERα-36
The antigenicity of the specific C-terminal amino
acid sequence of ERα-36 was analyzed using the Onastar software.
The IFGNKWFPRV sequence was selected and named IV10. This amino
acid sequence was made using solid phase chemical synthesis and
coupled by KLH. The specific antibody against IV10 was obtained
from the antiserum of rabbits and purified by Protein A affinity
chromatography.
Western blot analysis
MCF-7 and MDA-MB-231 breast cancer cells were
incubated with 20 ng/ml BMP2 for 24 and 48 h. For western blot
analyses, cells were disrupted by incubation at 4°C for 15 min in
cell lysis solution (UpState) containing protease inhibitor
cocktail (Roche). The protein concentration was quantified using a
BCA™ Protein Assay kit (Pierce). Equal amounts of protein (20 μg)
were subjected to 12% SDS-PAGE and western blot analysis, as
described previously (21).
Immunoreactive bands were visualised by an enhanced
chemiluminescence reaction kit (Thermo Fisher Scientific).
RNA interference
The sense strands of BMPR1a-siRNA and BMPR1b-siRNA
were purchased from Dharmacon, Inc. MDA-MB-231 cells were seeded
into 6-well plates, grown to 40–60% confluence and then transfected
with siRNAs for 4 h using Lipofectamine 2000 (Invitrogen). The
cells were allocated to 3 groups: control group, BMP2 group and
si-BMPR1a + si-BMPR1b + BMP2 group. The dosage of BMP2 was 20
ng/ml. The protein levels were analyzed by western blot
analysis.
Cell proliferation assay
For the proliferation assay with recombinant human
BMP2 (R&D Systems, Minneapolis, MN, USA), MCF-7 and MDA-MB-231
cells were cultured in 96-well plates (approximately 5,000 cells
per well) for 24 h, respectively. Cells were then serum-starved for
24 h in DMEM with 1% FBS. The experiment included a control group
and a BMP2 group (2.5, 5, 10, 20 or 30 ng/ml). After 48 h of
induction by BMP2, cell growth was measured by an MTT assay.
Tamoxifen citrate (Sigma) was used to identify the
correlation between BMP2 and 17-β-estradiol (E2) (Sigma). Cells
were plated in 96-well plates (approximately 5,000 cells/well) for
24 h in phenol red-free DMEM containing 2% FBS that had been
incubated with dextran-coated charcoal to remove endogenous
steroids (dialyzed fetal bovine serum, PAA Laboratories). Cells
were then incubated with BMP2 (20 ng/ml), E2 (0.01 μM) and
tamoxifen citrate (Tam, 0.01 μM) for 48 h. The experiment included
a control group, a BMP2 group (20 ng/ml), an E2 group, a Tam group,
a E2 + BMP2 group and a Tam + BMP2 group. The MTT assay was used to
determine the relative cell number. Absorbance was recorded at 570
nm in a spectrophotometer (Spectronic 1001, Bausch & Lomb). The
mean value of 5 wells was calculated and each experiment was
repeated 3 times.
Statistical analysis
Statistical analysis was carried out using the
Statistical Package for Social Sciences 13.0 (SPSS). Data are
presented as the means ± SEM. Statistical significance was
determined by a one-way analysis of variance or the t-test.
P-values <0.05 were considered to indicate statistically
significant differences.
Results
BMP2 alters the expression of genes in
MCF-7 and MDA-MB-231 cells
To understand the effect of BMP2 on the BMP and ER
signaling pathway in MCF-7 and MDA-MB-231 cells, the changes in the
expression of key genes, such as BMPR1a, BMPR1b, BMPR2, ERα and
ERβ, were examined using quantitative RT-PCR following the addition
of 20 ng/ml BMP2. Interestingly, we found that treatment with BMP2
upregulated the expression of ERα almost 7-fold in MCF-7 and 4-fold
in MDA-MB-231 cells. Since ERα had at least 3 types of identified
splicing variants (ERα-66, ERα-46 and ERα-36), we designed specific
primers for ERα-66 and ERα-36 and examined their specific
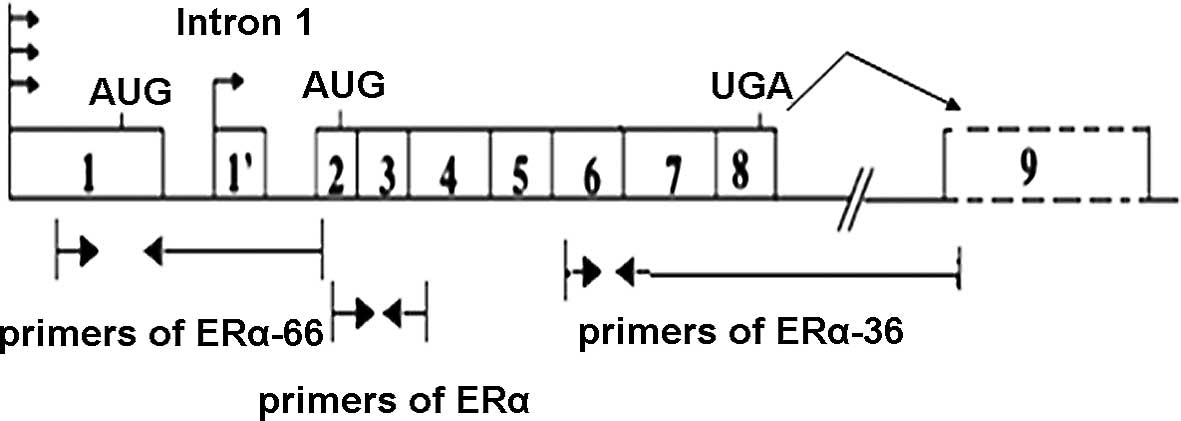
expression. Genomic organization of the human ERα-66/36 gene and
the positions of the primer pairs were shown in Fig. 1. The results shown in Fig. 2 indicated that the upregulation of
ERα-36 by BMP2 was sustained from 24 to 48 h in MCF-7 cells and was
the highest at 48 h in MDA-MB-231 cells. ERα-66 expressed in MCF-7
cells was slightly higher at 24 h. By contrast, the expression of
ERβ was not changed after the addition of BMP2 (Fig. 2) in either of the cell lines.
 | Figure 2Changes in ERα (including the 3
isoforms: 66, 46 and 36KD), ERα-66 and ERα-36 mRNA expression in
MCF-7 and MDA-MB-231 cells after induced by BMP2 (20 ng/ml). (A)
MCF-7 cells treated with BMP2. The expression of ERα was highest at
24 h, but dropped at 48 h. The expression of ERα-36 was upregulated
constantly from 24 to 48 h. (B) MDA-MB-231 cells treated with BMP2.
The expression of ERα and ERα-36 was highest at 48 h, and ERα-36
was upregulated 6-fold at 48 h. The value of 2–ΔΔCt
represents the expression of the ERα, ERα-66, ERα-36, BMPR1a,
BMPR1b and BMPR2 genes in BMP2-treated cells normalized to β-actin,
relative to the normalized expression of ERα, ERα-66, ERα-36,
BMPR1a, BMPR1b and BMPR2 genes in the control cells, respectively.
Results of 3 independent experiments were averaged and the mean
values ± SEM are shown. *p<0.05 compared to the 0 h
group. |
BMP2 induces the expression of ERα-36 in
MCF-7 and MDA-MB-231 cells
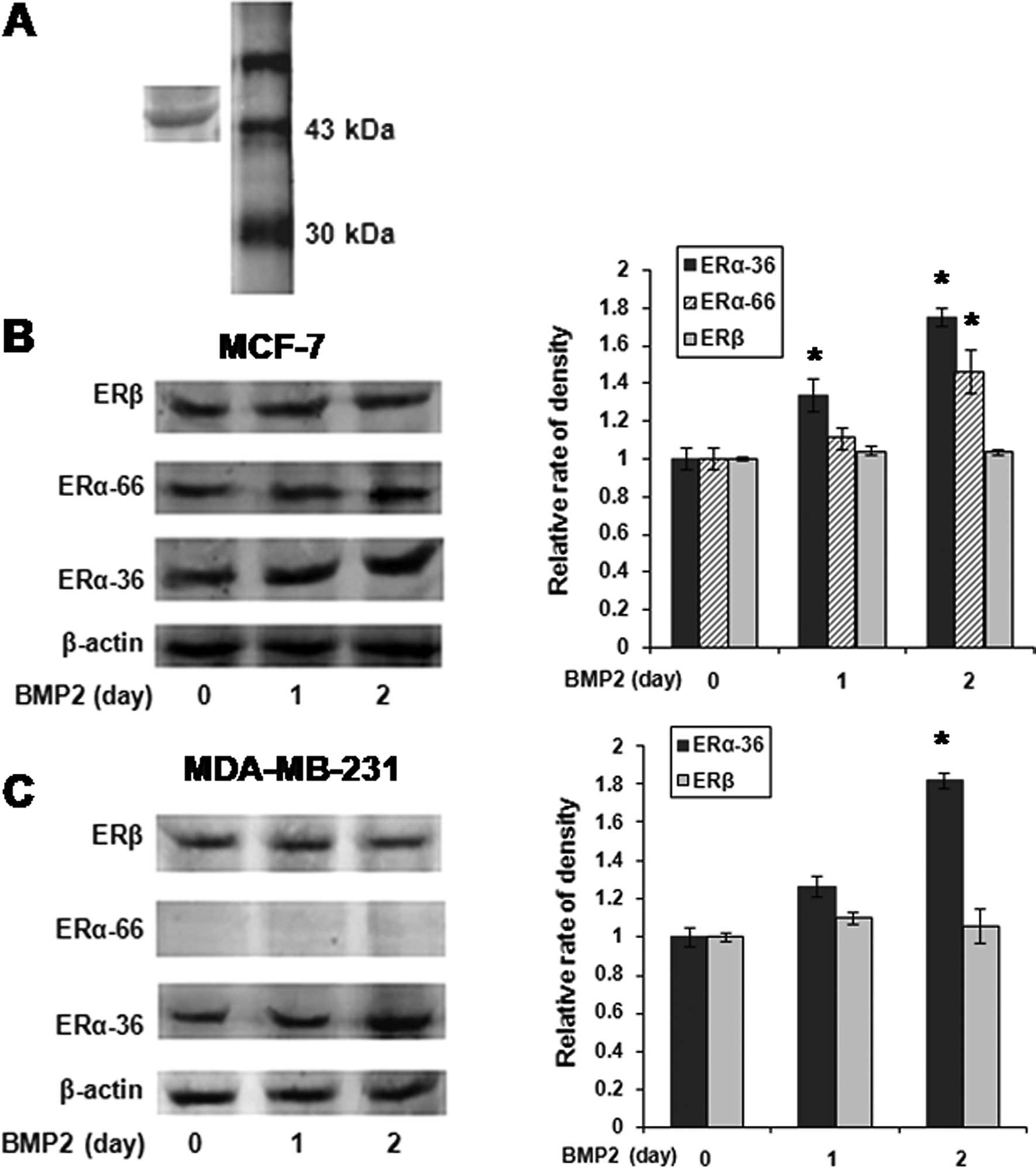
Western blot analysis was used to evaluate the
protein levels of ERα in BMP2-treated breast cancer cells. Since
the C-terminal of ERα-36 was unique, we raised antibodies against a
synthetic peptide antigen (IV10) corresponding to the C-terminal 10
aa of hERα-36. Its specificity was determined in MDA-MB-231 cells
(Fig. 3A). We observed that ERα-36
was significantly upregulated by BMP2 in MDA-MB-231 cells, which
were ERα-66-negative (Fig. 3B). We
also found that BMP2 induced the expression of ERα-36, but not
ERα-66 in MCF-7 cells (Fig. 3C).
The expression of ERβ did not change in either of the cell
lines.
BMP2 regulates the expression of the
ERα-36 protein
To assess the correlation between BMP2 and ERα-36,
an additional investigation was carried out on MDA-MB-231 cells
(ERα-66-negative). BMP2 induced the expression of ERα-36 in a
dose-dependent manner, which was inhibited by the BMP2 antagonist,
noggin (Fig. 4A). In addition, the
RNA interference assay indicated that BMP2 was associated with
ERα-36 expression. When the BMP2 signaling pathway was silenced by
si-BMPR1a and si-BMPR1b, the ERα-36 induction was eradicated
(Fig. 4B). Hence, it is possible
that crosstalk exists between BMP2 and ERα-36.
BMP2 inhibits the growth of MCF-7 and
MDA-MB-231 cells
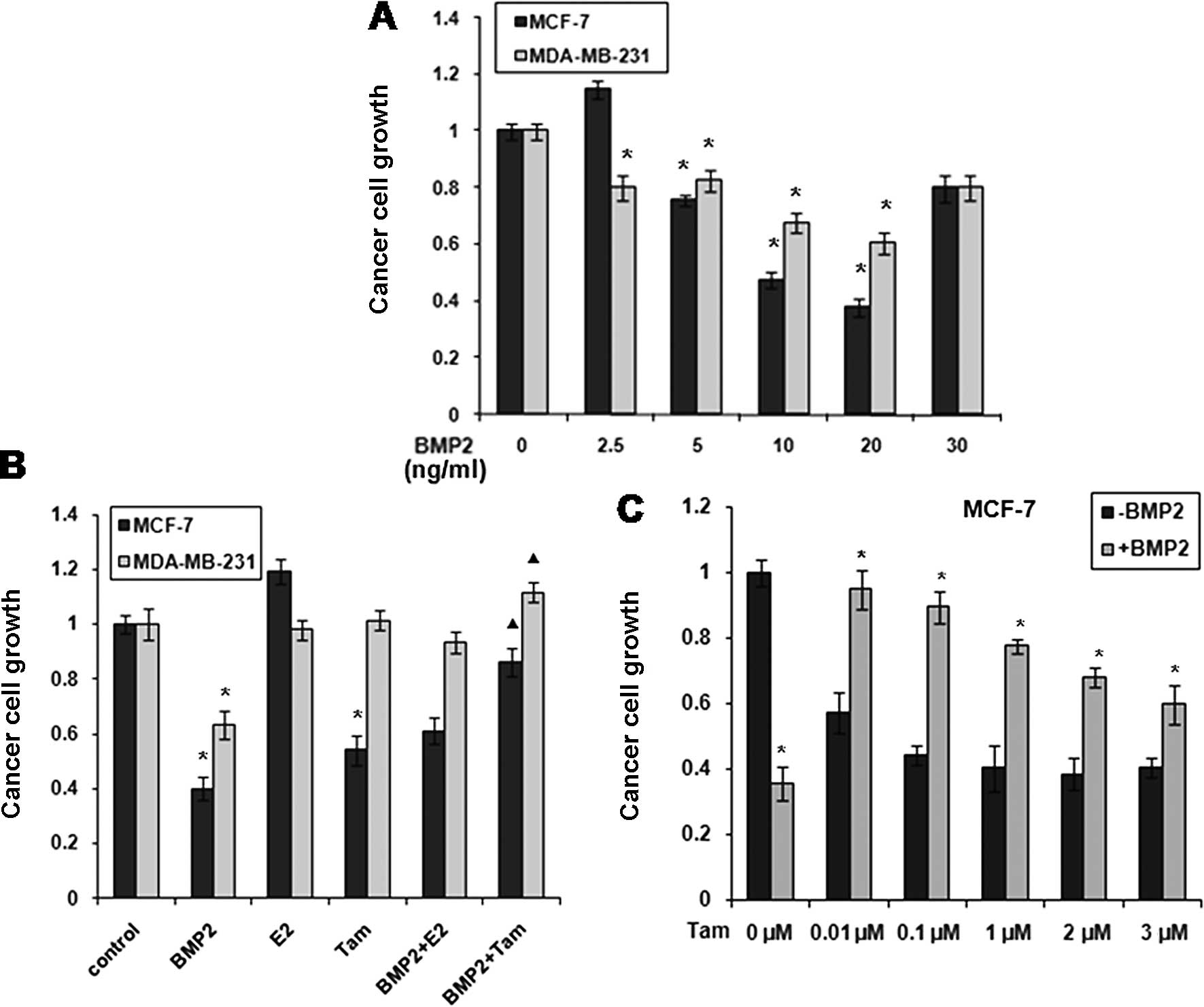
MTT assays revealed that the proliferation of MCF-7
and MDA-MB-231 cells was significantly inhibited by BMP2 (2.5, 5,
20 or 30 ng/ml) for 2 days. BMP-2 was effective in inhibiting the
growth of the MCF-7 cells when its dose was 5–30 ng/ml, but the
most effective dose was 20 ng/ml. A similar effect of BMP-2 was
observed in the MDA-MB-231 cells; however, its inhibitory effect on
cell proliferation was more prominent in MCF-7 than in MDA-MB-231
cells (Fig. 5A).
MDA-MB-231 cells were not sensitive to E2 and
tamoxifen treatment as shown in Fig.
2B. However, the BMP2 inhibition of MDA-MB-231 cells was
antagonised when E2 or tamoxifen were added. E2 promoted the
multiplication of MCF-7 cells, while tamoxifen restrained their
growth (Fig. 5B). The inhibitory
effect of BMP2 was, however, counteracted by tamoxifen. We also
observed that MCF-7 cells treated with BMP2 were insensitive to
tamoxifen treatment (Fig. 5C). The
results from our study were consistent with those from previous
reports stating that tamoxifen strongly inhibits cell proliferation
in the MCF-7 cells. The constitutive overexpression of recombinant
ERα-36, however, demonstrated insensitivity to tamoxifen treatment
(22). Since the MDA-MB-231 cells
were ERα-66-negative and ERα-36-positive following BMP2 treatment,
we hypothesized that the breast cancer cells became sensitive to E2
and resistant to tamoxifen following the induction of ERα-36 by
BMP2.
Discussion
In the present study, we identified that BMP2
induced the expression of ERα-36 in MDA-MB-231 and MCF-7 breast
cancer cells. A previous study on the induction of ERα and ERβ in
granulosa cells by activin also showed that BMP-2 increased ERα
mRNA levels by approximately 50% (23), consistent with our results.
Wang et al (21) reported that ERα-36 lacks both
transcriptional activation domains of ERα-66, retains portions of
the DNA-binding domain, the partial dimerization and ligand-binding
domains, and possesses a unique 27 amino acid domain that replaces
the last 138 amino acid of ERα-66. Moreover, ERα-36 can inhibit the
transactivation of both ERα-66 and ERβ, stimulate the MAPK
signaling pathway and induce cell growth in breast cancer cell
lines. Considering all the above information, the effect of ERα-36
on the growth inhibition induced by BMP2 is complicated, thus
additional investigation is required to clarify it.
An issue that scientists and clinical physicians
should address is why endocrine therapy is effective on some kinds
of breast cancers, but not on others. Recent studies have indicated
that the efficacy of endocrine therapy cannot be simply explained
by the expression levels of ERα, but may be determined by the
expression profiles of both ERα and ERβ, as well as their various
splice variants (14,21,24,25).
Furthermore, to understand the meaning of the demonstrated change
in ERα-36 expression induced by BMP2 in breast cancer cells,
additional research should be carried out to determine whether the
ERα-36 reported in the present study is the same as that reported
by Wang et al (21).
Anti-estrogen compounds are widely used for the
treatment of osteoporosis, breast cancer and other diseases. Smad4,
a common signal transducer in the BMP/TGF-β signaling pathway,
functions as a transcriptional co-repressor for human ERα (26). Estrogen and glucocorticoid interact
in osteoblastic differentiation regulated by BMP and TNF-α in mouse
myoblastic C2C12 cells. The expression of ERs, ERα and ERβ, as well
as the glucocorticoid receptor (GCR) has been shown to be
significantly increased by BMP-2 treatment, regardless of the
presence of estradiol and dexamethasone (27). ERα, BMP-2 and BMP-4 expressions
have been found to correlate temporarily, during the development of
the osteoblast phenotype in early fibroblastic stem cells (28). These observations indicate that the
ER pathway is closely associated with the BMP pathway during bone
development and remodeling. In previous studies, BMP-6 expression
was determined to be activated by estrogen in the MCF-7 breast
cancer cell line (29). It was
also reported that BMP-2-induced the activation of Smad activity
and that BMP-2-mediated gene expression was suppressed by E2 in
breast cancer cells via direct physical interactions between Smads
and ER (30). Therefore, the
crosstalk between the BMP and ER pathways may play a role in tumor
differentiation and metastasis. It is reported in this study, for
the first time, that BMP2 can induce the expression of ERα-36 in
MCF-7 and MDA-MB-231 cells. This study provides new insights into
the BMP2 function in breast cancer cells. Understanding the
crosstalk of BMP2 and ER signaling pathways will increase our
understanding of the mechanisms of many diseases, in addition to
cancer, that are important to human health.
Acknowledgements
The authors would like to thank Miss Li Yueqin for
the technical support and Drs Li Hongjian and Cai Dongqing for
having discussed the key issues in this study. The present study
was supported by grants from the Guangzhou Scientific and Technical
Project (33107005) and by the Foundation for Distinguished Young
Talents in Higher Education of Guangdong, China (LYM11080).
References
|
1
|
Miyazono K, Kamiya Y and Morikawa M: Bone
morphogenetic protein receptors and signal transduction. J Biochem.
147:35–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wozney JM, Rosen V, Celeste AJ, Mitsock
LM, Whitters MJ, Kriz RW, Hewick RM and Wang EA: Novel regulators
of bone formation: molecular clones and activities. Science.
242:1528–1534. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Attisano L and Wrana JL: Signal
transduction by the TGF-beta superfamily. Science. 296:1646–1647.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee J, Son MJ, Woolard K, Donin NM, Li A,
Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, et al:
Epigenetic-mediated dysfunction of the bone morphogenetic protein
pathway inhibits differentiation of glioblastoma-initiating cells.
Cancer Cell. 13:69–80. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Varga AC and Wrana JL: The disparate role
of BMP in stem cell biology. Oncogene. 24:5713–5721. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Farnsworth RH, Karnezis T, Shayan R,
Matsumoto M, Nowell CJ, Achen MG and Stacker SA: A role for bone
morphogenetic protein-4 in lymph node vascular remodeling and
primary tumor growth. Cancer Res. 71:6547–6557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Katzenellenbogen BS and Katzenellenbogen
JA: Estrogen receptor transcription and transactivation: Estrogen
receptor alpha and estrogen receptor beta: regulation by selective
estrogen receptor modulators and importance in breast cancer.
Breast Cancer Res. 2:335–344. 2000. View
Article : Google Scholar
|
|
8
|
Glidewell-Kenney C, Weiss J, Lee EJ,
Pillai S, Ishikawa T, Ariazi EA and Jameson JL: ERE-independent
ERalpha target genes differentially expressed in human breast
tumors. Mol Cell Endocrinol. 245:53–59. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Holmes KA, Hurtado A, Brown GD, Launchbury
R, Ross-Innes CS, Hadfield J, Odom DT and Carroll JS:
Transducin-like enhancer protein 1 mediates estrogen receptor
binding and transcriptional activity in breast cancer cells. Proc
Natl Acad Sci USA. 109:2748–2753. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vasudevan N, Kow LM and Pfaff DW: Early
membrane estrogenic effects required for full expression of slower
genomic actions in a nerve cell line. Proc Natl Acad Sci USA.
98:12267–12271. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Baselga J, Semiglazov V, van Dam P,
Manikhas A, Bellet M, Mayordomo J, Campone M, Kubista E, Greil R,
Bianchi G, et al: Phase II randomized study of neoadjuvant
everolimus plus letrozole compared with placebo plus letrozole in
patients with estrogen receptor-positive breast cancer. J Clin
Oncol. 27:2630–2637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Edwards DP: Regulation of signal
transduction pathways by estrogen and progesterone. Annu Rev
Physiol. 67:335–376. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu H, Qiu J, Li N, Chen T and Cao X:
Human phosphatidylethanolamine-binding protein 4 promotes
transactivation of estrogen receptor alpha (ERalpha) in human
cancer cells by inhibiting proteasome-dependent ERalpha degradation
via association with Src. J Biol Chem. 285:21934–21942. 2010.
View Article : Google Scholar
|
|
14
|
Thomas C and Gustafsson JÅ: The different
roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer.
11:597–608. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herynk MH and Fuqua SA: Estrogen receptor
mutations in human disease. Endocr Rev. 25:869–898. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Z, Zhang X, Shen P, Loggie BW, Chang
Y and Deuel TF: Identification, cloning, and expression of human
estrogen receptor-alpha36, a novel variant of human estrogen
receptor-alpha66. Biochem Biophys Res Commun. 336:1023–1027. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Osborne CK: Tamoxifen in the treatment of
breast cancer. N Engl J Med. 339:1609–1618. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miller TW, Balko JM and Arteaga CL:
Phosphatidylinositol 3-kinase and antiestrogen resistance in breast
cancer. J Clin Oncol. 29:4452–4461. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ho CC and Bernard DJ: Bone morphogenetic
protein 2 acts via inhibitor of DNA binding proteins to
synergistically regulate follicle-stimulating hormone β
transcription with activin A. Endocrinology. 151:3445–3453.
2010.PubMed/NCBI
|
|
20
|
ten Dijke P, Korchynskyi O,
Valdimarsdottir G and Goumans MJ: Controlling cell fate by bone
morphogenetic protein receptors. Mol Cell Endocrinol. 211:105–113.
2003.PubMed/NCBI
|
|
21
|
Wang Z, Zhang X, Shen P, Loggie BW, Chang
Y and Deuel TF: A variant of estrogen receptor-{alpha},
hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent
membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA.
103:9063–9068. 2006.
|
|
22
|
Lin SL, Yan LY, Zhang XT, et al:
ERalpha-36, a variant of ER-alpha, promotes tamoxifen agonist
action in endometrial cancer cells via the MAPK/ERK and PI3K/Akt
pathways. PLoS One. 5:e90132010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kipp JL, Kilen SM, Woodruff TK and Mayo
KE: Activin regulates estrogen receptor gene expression in the
mouse ovary. J Biol Chem. 282:36755–36765. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J,
Wang T, Fan Z, Fan T, Lin B, Wang Z and Xie Y: Expression of
ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and
resistance to tamoxifen treatment in breast cancer. J Clin Oncol.
27:3423–3429. 2009.
|
|
25
|
Lee LM, Cao J, Deng H, Chen P, Gatalica Z
and Wang ZY: ER-alpha36, a novel variant of ER-alpha, is expressed
in ER-positive and -negative human breast carcinomas. Anticancer
Res. 28:479–483. 2008.PubMed/NCBI
|
|
26
|
Wu L, Wu Y, Gathings B, Wan M, Li X,
Grizzle W, Liu Z, Lu C, Mao Z and Cao X: Smad4 as a transcription
corepressor for estrogen receptor alpha. J Biol Chem.
278:15192–15200. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matsumoto Y, Otsuka F, Takano M, Mukai T,
Yamanaka R, Takeda M, Miyoshi T, Inagaki K, Sada KE and Makino H:
Estrogen and glucocorticoid regulate osteoblast differentiation
through the interaction of bone morphogenetic protein-2 and tumor
necrosis factor-alpha in C2C12 cells. Mol Cell Endocrinol.
325:118–127. 2010. View Article : Google Scholar
|
|
28
|
Oreffo RO, Kusec V, Romberg S and Triffitt
JT: Human bone marrow osteoprogenitors express estrogen
receptor-alpha and bone morphogenetic proteins 2 and 4 mRNA during
osteoblastic differentiation. J Cell Biochem. 75:382–392. 1999.
View Article : Google Scholar
|
|
29
|
Zhang M, Wang Q, Yuan W, Yang S, Wang X,
Yan JD, Du J, Yin J, Gao SY, Sun BC and Zhu TH: Epigenetic
regulation of bone morphogenetic protein-6 gene expression in
breast cancer cells. J Steroid Biochem Mol Biol. 105:91–97. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamamoto T, Saatcioglu F and Matsuda T:
Cross-talk between bone morphogenic proteins and estrogen receptor
signaling. Endocrinology. 143:2635–2642. 2002. View Article : Google Scholar
|