Introduction
Multiple myeloma (MM) is a plasma cell malignancy
and the second most common hematological malignancy after
non-Hodgkin lymphoma (1). Despite
advances in supportive and systemic therapies, MM remains an
essentially incurable disease with a high rate of relapse and a
rapid acquisition of drug resistance. The development of MM is a
multistep process associated with an increasing frequency of
genetic aberrations, including chromosomal abnormalities, as well
as complex translocations, including the complete deletion of
chromosome 13 or its long arm. In addition, increased density of
bone marrow (BM) microvessels is associated with MM development
(2–4).
However, an increasing number of studies are
reporting that, in addition to genetic abnormalities, epigenetic
processes play a major role in the carcinogenesis of MM (5,6). DNA
methylation of cytosine bases within a CpG dinucleotide represents
a major epigenetic modification that results in altered
transcriptional activity in the molecular pathogenesis of various
hematological malignancies (7). In
MM, numerous cases of epigenetic silencing of tumor suppressor
genes (TSGs), including p15, p16, DAP-kinase, CDH1, DLC1, SOCS-1
and E-Cadherin, have been reported (8–11).
In addition, a member of the a disintegrin-like and metalloprotease
with thrombospondin type I motifs (ADAMTS) family has been
suggestedc to be involved in processes regulating epigenetic gene
silencing in MM.
The ADAMTS superfamily is involved in a wide range
of cellular processes, including the maturation of procollagen and
the extracellular matrix proteolysis associated with morphogenesis,
cancer, angiogenesis, arthritis and ovulation. Several members of
the ADAMTS family (ADAMTS9, ADAMTS12, ADAMTS15 and ADAMTS18) have
been reported as candidate TSGs.
ADAMTS9, which is located at 3p14.2, is the most
highly conserved member of this superfamily. ADAMTS9 is involved in
a number of biological processes, including melanoblast
development, vascular development and the suppression of
angiogenesis in the tumor context (12). The gene functions as a TSG via
promoter methylation in various types of human cancer, such as
esophageal, nasopharyngeal, gastric, colorectal and pancreatic
(13–15).
In the present study, the expression and promoter
methylation status of ADAMTS9 was analyzed in MM cell lines and
patients. Promoter hypermethylation and the re-expression of
ADAMTS9 were performed using demethylating drug treatment and
results revealed that promoter methylation is the key mechanism of
ADAMTS9 expression inactivation in MM. Ectopic ADAMTS9 expression
in KM3 cells leads to the significant suppression of colony
formation and cell proliferation, indicating that ADAMTS9 is a
novel functional tumor suppressor gene.
Materials and methods
Cell culture and clinical samples
KM3 and RPMI-8226 cell lines were obtained from Dr
Jian Hou (The Second Military Medical University, Shanghai, China)
and maintained in RPMI-1640 medium supplemented with 10% fetal
bovine serum (both Gibco-BRL, Carlsbad, CA, USA). A total of 32
male and 24 female patients with a median age of 64 (range, 38–78)
years, recently diagnosed with MM, as well as 16 healthy adults,
who served as controls, were included in the study. Patients were
previously untreated. BM samples were obtained from the Hematology
Laboratory of The First Affiliated Hospital of Chongqing Medical
University (Chongqing, China). The diagnosis of MM was based on
standard criteria (International Myeloma Working Group, 2003) and
the patients were classified according to the International Staging
System (ISS). The samples were confirmed to have no solid tumor
infiltration. The study was conducted according to the principles
of the Declaration of Helsinki and informed consent was obtained
from all patients.
Methyltransferase inhibitor drug
treatment
Cells were treated with 10 μM 5-aza-2′-deoxycytidine
(Aza; Sigma-Aldrich, St. Louis, MO, USA) for 3 days and then with
100 ng/ml trichostatin A (TSA; Sigma-Aldrich) for an additional 24
h, as described previously (16).
Total RNA isolation and semi-quantitative
reverse transcription PCR (RT-PCR)
RNA was extracted using TRIzol reagent (Invitrogen
Life Technologies, Carlsbad, CA, USA) and reverse-transcribed using
an RT reagent kit (Takara Bio, Inc., Shiga, Japan) and random
hexamer primers. PCR analysis using Go-Tag (Promega Corporation,
Madison, WI, USA) was perfomed using GAPDH as a control. ADAMTS9
expression was analyzed by PCR using the following primers:
ADAMTS9, F: 5′-CAT GCA GTT TGT ATC CTG-3′ and R: 5′-GCG TTC TTT TGA
AGT GGA CG -3′; GAPDH, F: 5′-ATC TCT GCC CCC TCT GCT GA-3′ and R:
5′-GAT GAC CTT GCC CAC AGC CT-3′. RT-PCR was performed with 32
cycles for ADAMTS9 and 23 cycles for GAPDH.
DNA bisulfite treatment and
methylation-specific polymerase chain reaction (MSP)
Genomic DNA was extracted using the QIAmp DNA blood
Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's
instructions. Bisulfite modification of DNA and methylation of the
CpG islands of the ADAMTS9 promoter were performed as described
previously (13). The primers
detecting the methylated or unmethylated alleles of the ADAMTS9
promoter were as follows: ADAMTS9-m1: 5′-TTT TTC GTT TTT TTT TGT
TCG TTC-3′ and -m2: 5′-AAA CTA AAC CGC TCG AAC CG-3′ for the
methylated alleles and ADAMTS9-u1: 5′-GTT TTT TGT TTT TTT TTG TTT
GTT T-3′ and -u2: 5′-AAA AAC TAA ACC ACT CAA ACC A-3′ for the
unmethylated alleles. MSP was performed for 40 cycles using Ampli
Taq-Gold under methylation- and unmethylation-specific primer
conditions: annealing temperature 60 and 58°C, respectively. The
MSP primers were tested previously to ensure that DNA that was not
modified by bisulfite was not amplified.
Cell transfection
Transfection plasmids were purified using the
TIANprep Mini Plasmid kit [Tiangen Biotech (Beijing) Co., Ltd.,
Beijing, China]. KM3 cells (2×105 cells/well) were
plated in 6-well plates and transfected with the expression plasmid
(2 μg), pCEP4-ADAMTS9 or the empty vector (2 μg), pCEP4, using
Lipofectamine 2000 (Invitrogen Life Technologies) according to the
manufacturer's instructions. Cells were collected and plated in a
5-cm dish 48 h post-transfection and selected for 21 days with G418
(0.4 mg/ml).
Colony formation assay
Colony formation assay was performed using
semi-solid medium. Cells were suspended in RPMI-1640 medium
containing 1% methylcellulose, 35% fetal bovine serum and 0.8 mg/ml
G418 in a 5-cm dish 48 h post-transfection. The dish was placed in
a sealed chamber and incubated at 37°C in a 5% CO2
incubator for 21 days. The number of surviving colonies (≥50
cells/colony)/cm3 was quantified using an inverted
microscope. Total RNA from the transfected cells was extracted and
analyzed via RT-PCR to confirm the ectopic expression of ADAMTS9.
The experiments were performed in triplicate wells 3 times.
Flow cytometric analysis of the cell
cycle
KM3 cells were plated in 6-well plates and
transfected with 4 μg pCEP4-ADAMTS9 or the empty vector, pCEP4,
using Lipofectamine 2000 according to the manufacturer's
instructions. Stable ADAMTS9 expression and vector-KM3 cells were
harvested and fixed in ice-cold 70% ethanol for 1 h. Cell cycle
profiles were assayed using an Elite ESP flow cytometer and the
data were analyzed using CellQuest software (BD Biosciences,
Franklin Lakes, NJ, USA).
Measurement of cell proliferation
The impact of the ectopic expression of ADAMTS9 on
KM3 cell proliferation was assessed using the Cell Counting kit-8
(CCK-8) (Beyotime Institute of Biotechnology, Jiangsu, China)
according to the manufacturer's instructions. Briefly, KM3 cells
were transfected as described above and cultured at a density of
3×103 cells/well in 96-well plates. Cell proliferation
was monitored at 24, 48 and 72 h, 20 μl CCK-8 solution was added to
each well following 1 h incubation in a 5% CO2
humidified incubator at 37°C and the optical density was measured
at 450 nm using a microplate reader (Molecular Devices LLC,
Sunnyvale, CA, USA). Experiments were performed in 5
wells/experiment and repeated 3 times.
Statistical analysis
Statistical analyses were performed using SAS
version 9.1 for windows (SAS Institute Inc., Cary, NC, USA). Data
were presented as the mean ± SD. Associations between the ADAMTS9
methylation status of the tumor and non-tumor samples and the
clinical characteristics were assessed using Chi-square, Fisher's
exact and Student's t-tests. Differences between the subgroups
(colony formation assay, cell cycle analysis and proliferation
measurement) were assessed using the Student's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
ADAMTS9 was expressed in healthy normal
adult tissues and frequently silenced in MM cell lines and patient
samples
Expression levels of ADAMTS9 were analyzed in KM3
and RPMI-8226 cells, 16 MM BM samples and 16 normal adult BM
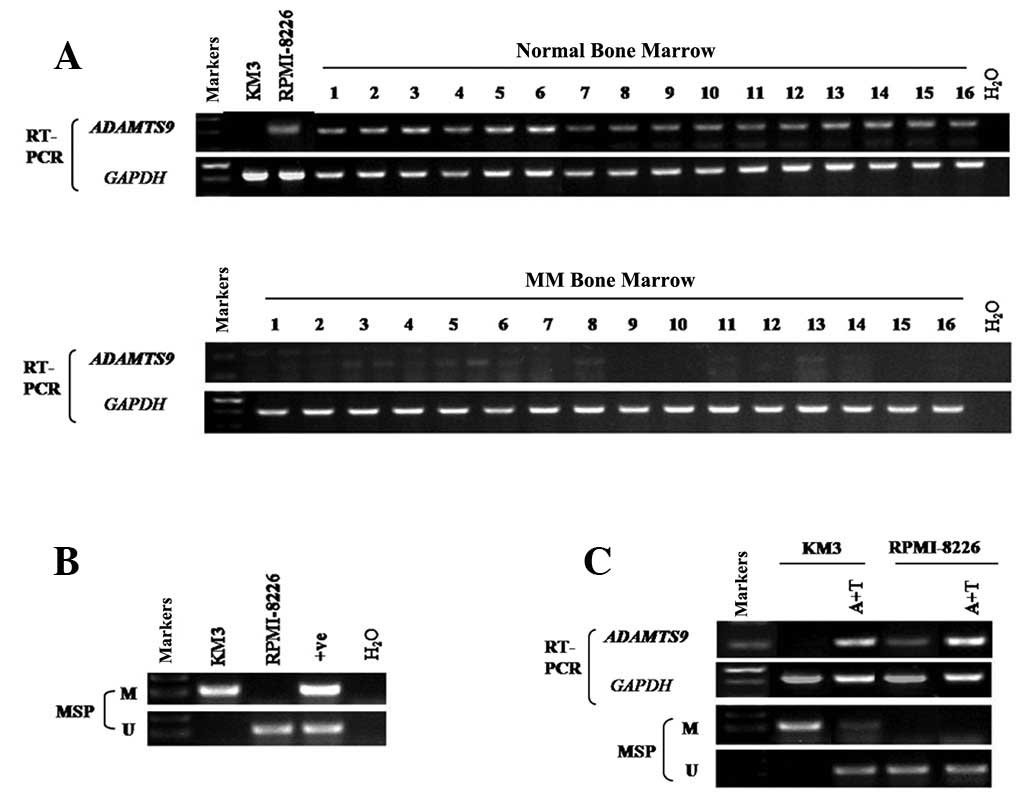
samples with semi-quantitative RT-PCR. The results revealed that
ADAMTS9 was silenced in 100% (16/16) of the MM samples (Fig. 1A), while its expression was readily
detected in the normal adult BM samples (0/16) (Fig. 1A). In addition, among the two MM
cell lines tested, KM3 revealed no ADAMTS9 expression (Fig. 1A). By contrast, ADAMTS9 expression
was detected in RPMI-8226 cells (Fig.
1A).
Frequent silencing of ADAMTS9 in MM cell
lines due to promoter methylation
As aberrant promoter CpG methylation is associated
with gene silencing, the methylation status of the MM cell lines
was determined using MSP. ADAMTS9 was methylated in KM3 (Fig. 1B), while methylation was not
observed in RPMI-8226 cells and normal adult BM (Figs. 1B and 2), consistent with the hypothesis that
ADAMTS9 methylation is negatively correlated with ADAMTS9
expression levels.
Activation of ADAMTS9 expression via
pharmacological demethylation
To examine whether promoter methylation of ADAMTS9
directly mediates expression silencing, cells were treated with the
DNA methyltransferase inhibitor, Aza, combined with the histone
deacetylase inhibitor, TSA. ADAMTS9 expression was markedly induced
following drug treatment in KM3 cells (Fig. 1C), accompanied with a decrease in
methylated alleles and an increase in unmethylated alleles of the
ADAMTS9 promoter in KM3 cells (Fig.
1C). However, no difference was observed in RPMI-8226 cells
(Fig. 1C). The results indicate
that DNA methylation of the ADAMTS9 promoter is directly involved
in transcriptional silencing in MM cells.
Frequent ADAMTS9 methylation and
clinicopathological correlations in MM
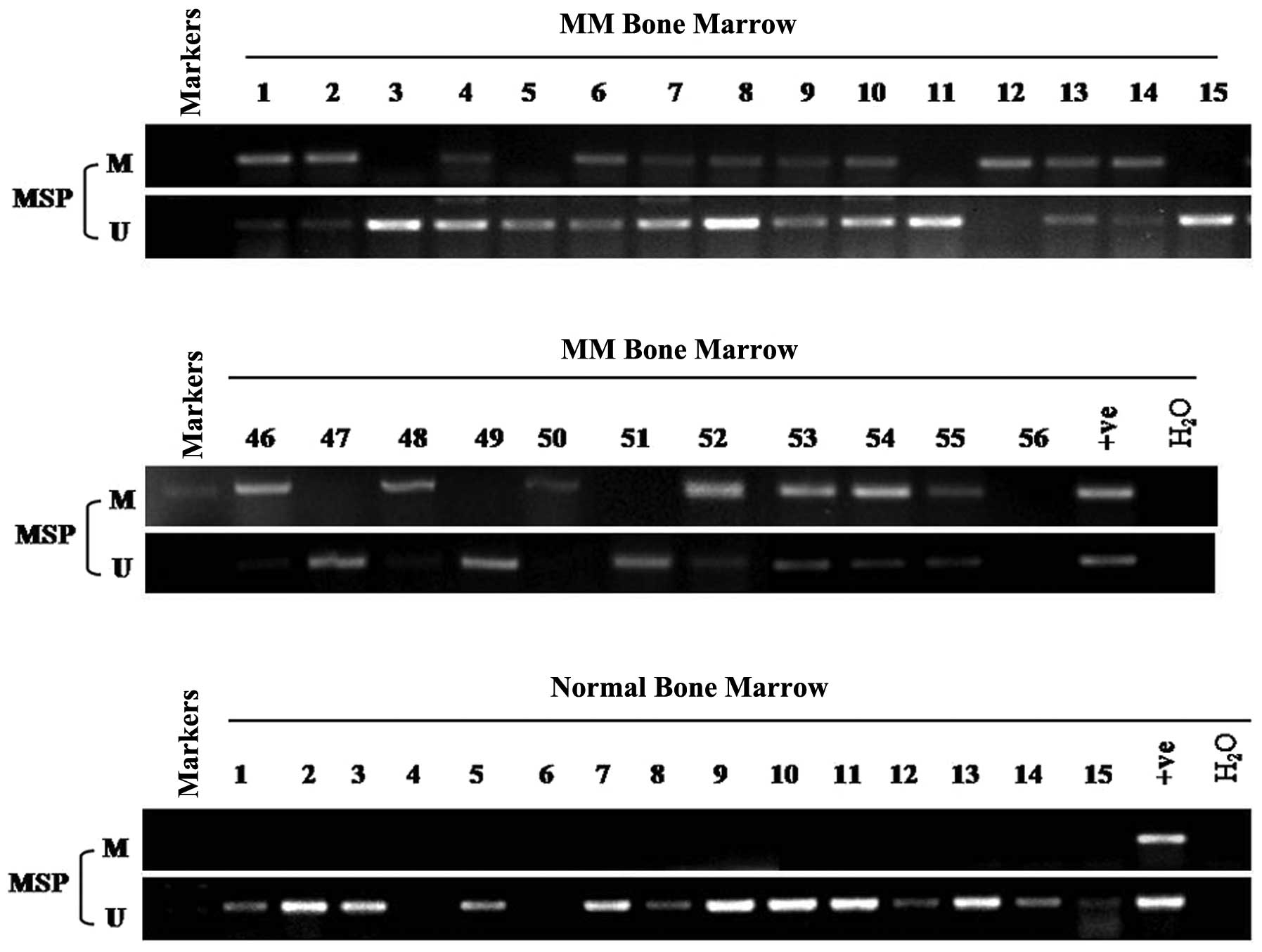
MSP was used to analyze the methylation status of
ADAMTS9 in 56 MM and 15 normal adult BM samples. Aberrant promoter
methylation was detected in 66% (37/56) of the MM BM samples
(Table I, Fig. 2), while no methylation was detected
in any of the 15 normal adult samples (Fig. 2). Potential correlations between
the ADAMTS9 methylation status and clinical parameters were
determined, including gender, age, MM subtype, ISS staging system
(Table II). However, no
significant correlations were observed between the patients with
methylated ADAMTS9 and these clinicopathological
characteristics.
 | Table IMethylation status of ADAMTS9 promoter
in MM (n=56). |
Table I
Methylation status of ADAMTS9 promoter
in MM (n=56).
| ADAMTS9 promoter | |
|---|
|
| |
|---|
| Samples | Methylated | Unmethylated | Frequency of
methylation (%) |
|---|
| Multiple myeloma,
n | 37 | 19 | 37/56 (66) |
| Table IICorrelations between ADAMTS9 promoter
methylation and clinicopathological indices of MM patients
(n=56). |
Table II
Correlations between ADAMTS9 promoter
methylation and clinicopathological indices of MM patients
(n=56).
| ADAMTS9 methylation
status | | |
|---|
|
| | |
|---|
| Clinical
parameters | Methylated
(n=37) | Unmethylated
(n=19) | Total | P-value |
|---|
| Gender | | | | 0.7144 |
| Male | 22 | 10 | 32 | |
| Female | 15 | 9 | 24 | |
| Age (years) | | | | 0.4612 |
| <65 | 16 | 16 | 32 | |
| ≥65 | 21 | 3 | 24 | |
| MM type | | | | 0.9897 |
| IgG | 16 | 7 | 23 | |
| IgA | 10 | 6 | 16 | |
| κ | 7 | 3 | 10 | |
| λ | 2 | 2 | 4 | |
| Non-secretory | 2 | 1 | 3 | |
| ISS stage | | | | 0.9247 |
| I | 12 | 7 | 19 | |
| II | 11 | 5 | 16 | |
| III | 14 | 7 | 21 | |
| White blood cell
(x109) | 5.42±2.47 | 5.96±1.81 | | 0.3947 |
| Hemoglobin
(g/l) | 96.59±21.10 | 91.85±25.46 | | 0.4546 |
| Platelets
(x109) | 166.12±75.75 | 191.74±93.50 | | 0.2739 |
| Serum calcium
(mmol/l) | 2.30±0.18 | 2.28±0.14 | | 0.6860 |
| Serum creatinine
(μmol/l) | 84.70±10.26 | 83.85±11.65 | | 0.7734 |
Ectopic expression of ADAMTS9 suppresses
tumor cell clonogenicity
The frequent silencing of ADAMTS9 in MM cell lines
compared with its broad expression in normal tissues indicates that
ADAMTS9 may exert tumor suppressor functions in this tumor type. To
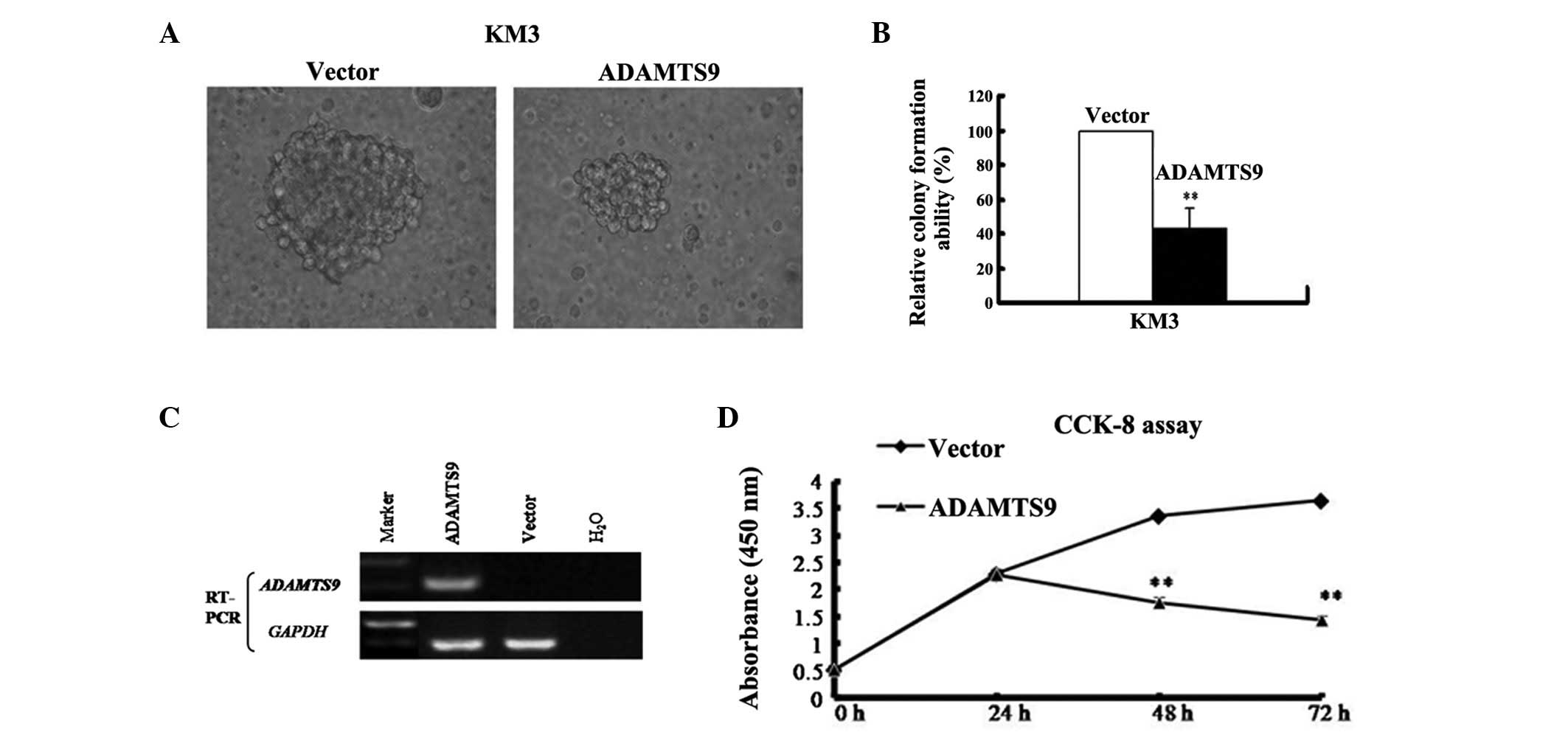
verify this hypothesis, a colony formation assay was performed to
test for the growth-inhibitory effect of ADAMTS9 in KM3 cells with
complete methylation and silencing of ADAMTS9 (Fig. 3A). The ADAMTS9-expressing and empty
vector (control) were transfected into KM3 cells. The cells were
then selected with 400 μg/ml G418 (Merck, Darmstadt, Germany) to
obtain stably transfected cells. Following G418 selection for 2
weeks, ADAMTS9 was stably overexpressed as revealed by RT-PCR
(Fig. 3C). A sharp reduction in
colonies (40–60% of the control vector; P<0.01) was observed in
the KM3-ADAMTS9 cells compared with the empty vector control
(Fig. 3B). These results indicate
that ADAMTS9 suppresses colony formation and may act as a tumor
suppressor in MM cells.
Ectopic expression of ADAMTS9 inhibits
tumor cell proliferation and induces S-phase cell cycle arrest
The CCK-8 assay was employed to examine the effect
of ADAMTS9 on MM cell proliferation in vitro. The growth of
ADAMTS9- and vector-expressing KM3 cells was observed at 0, 24, 48
and 72 h. The proliferation trend was identified to be
significantly decreased in ADAMTS9-expressing KM3 cells at 48 and
72 h (P<0.01) but increased in the vector-expressing KM3 cells
in a time-dependent manner (Fig.
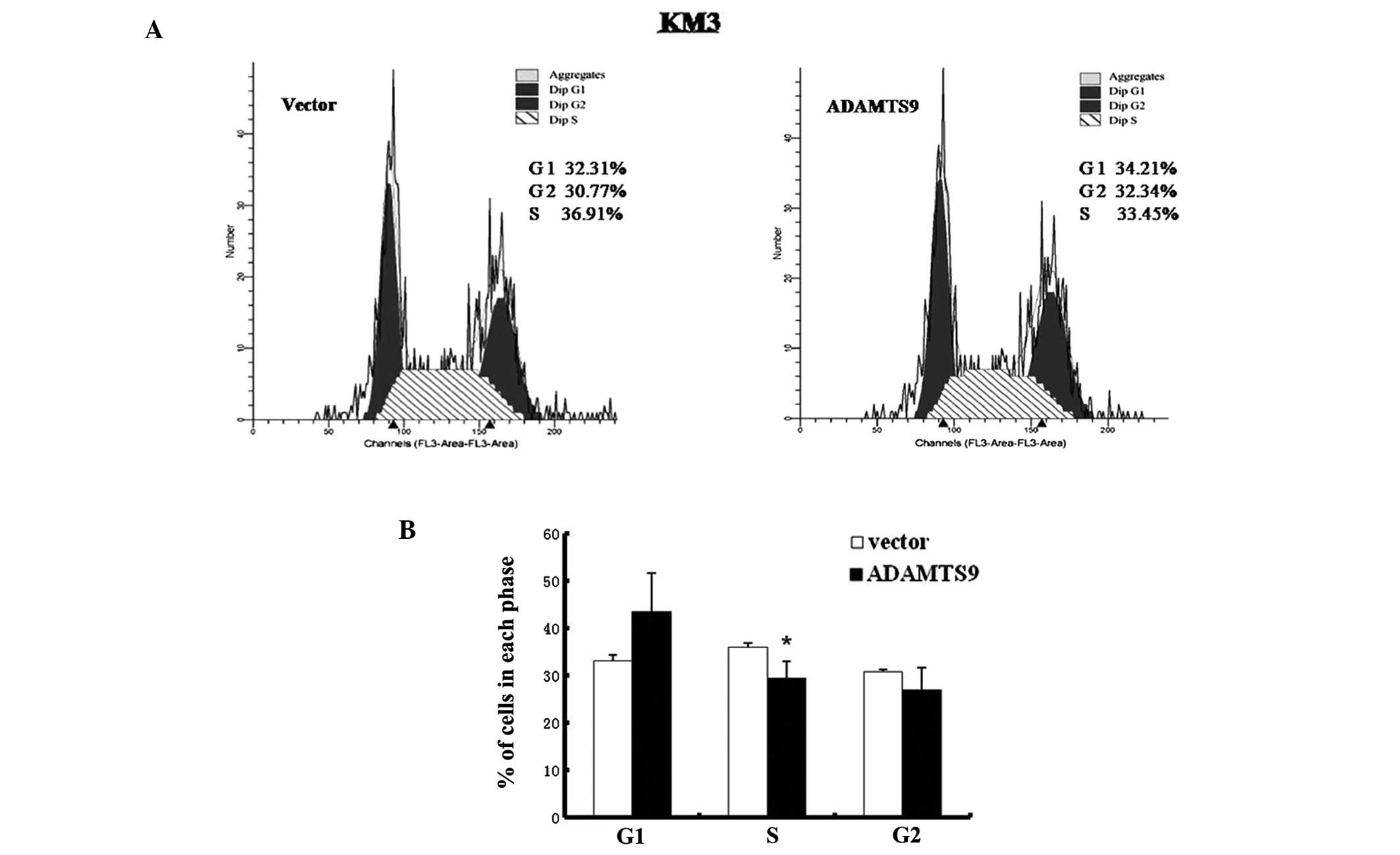
3D). To examine the mechanism by which ADAMTS9 inhibits the
proliferation of KM3 cells in vitro, the effect of ADAMTS9
expression on cell cycle distribution was investigated using flow
cytometry (Fig. 4A). As
demonstrated in Fig. 4B, the
proportion of cells in the S phase decreased significantly in
ADAMTS9-transfected KM3 cells, which was accompanied by an increase
in cells in the G1 phase compared with the empty
vector-transfected KM3 cells (P<0.05). These observations are
consistent with the hypothesis that the inhibitory effect of cell
proliferation induced by ADAMTS9 expression is mediated by cell
cycle arrest in the S phase.
Discussion
Aberrant promoter methylation may be important in
the carcinogenic process (17).
Previously, a number of genes, including PTGS2, SFN, CDKN2B, CDH1,
ESR1, HIC1, CCND2 and TGFβR2, were found to be silenced in
association with aberrant methylation in MM (18). Analysis of esophageal and
nasopharyngeal cancer demonstrated that the aberrant methylation of
ADAMTS9 is important for malignancy (13). However, ADAMTS9 methylation has not
been detected in MM.
The present study confirms that ADAMTS9 is
abundantly expressed in normal adult BM samples and RPMI-8226 cells
but silenced in KM3 cells and MM BM samples, thereby indicating
that ADAMTS9 plays a vital role in the pathogenesis of MM. This
study revealed that promoter methylation of ADAMTS9 occurs in KM3
cells and in 66% of MM samples. These observations indicate that
the disruption of ADAMTS9 in MM is markedly associated with
aberrant promoter methylation. However, methylation was not
detected in RPMI-8226 cells. This demonstrates that methylation of
ADAMTS9 is not the only cause of MM and the involvement of
additional mechanisms cannot be excluded. Therefore, further
studies are required to understand the mechanism of MM
development.
Previously, ADAMTS9 expression was found to
significantly correlate with lymph node metastases in
nasopharyngeal carcinoma (14).
However, a significant association was not observed between the
methylation status of ADAMTS9 and the clinicopathological indices
of our 56 MM patients, indicating that ADAMTS9 methylation may be
an early event in MM tumorigenesis. However, the clinical impact of
ADAMTS9 inactivation via promoter methylation in MM remains
unknown. To assess the correlation between methylation levels of
ADAMTS9 with the clinical parameters, studies with larger cohorts,
as well as more potential diagnostic and prognostic clinical
features are required. ADAMTS9 promoter methylation was detected at
a high frequency in MM tissues but not in normal tissues, thereby
demonstrating that ADAMTS9 may be a potential cancer-specific
biomarker for MM diagnosis. To the best of our knowledge, this is
the first study to identify that ADAMTS9 is epigenetically silenced
in human MM.
Silencing of ADAMTS9 may be activated by
pharmacological demethylation, suggesting that aberrant promoter
methylation is a mechanism for ADAMTS9 silencing in MM cells. In
the current study, colony-formation, flow cytometry and CCK-8 assay
results revealed that as a tumor suppressor, ADAMTS9 suppresses
tumor cell clonogenicity and cell proliferation, which may be
mediated by cell cycle arrest at the S phase following ADAMTS9
re-expression. Lung et al(14) demonstrated that a marked reduction
in the colony-forming ability was observed following transfection
of ADAMTS9 into NPC cell lines, which is consistent with our
study.
ADAMTS9, together with other members of the ADAMTS
family, is involved in tumorigenesis (19–23),
which may be associated with anti-angiogenic activity (13, 24–26).
Previously, ADAMTS9 was revealed to be a cell-autonomous
angiogenesis inhibitor (27). In
addition, ADAMTS9 has been identified to function in angiogenesis,
however, its role in MM tunorigenesis remains unclear. Further
analyses must be conducted to improve understanding of the
underlying mechanism.
In conclusion, ADAMTS9 was identified to be
frequently silenced by promoter methylation in MM in a
cancer-specific manner, which may be reversed by treatment with a
demethylation reagent. The transfection of ADAMTS9 into MM cells
lacking ADAMTS9 expression led to a reduction in cell colony
forming ability and cell proliferation mediated by cell cycle
arrest in the S phase, indicating that epigenetic inactivation of
ADAMTS9 is an important factor in MM carcinogenesis. ADAMTS9 may be
a candidate tumor suppressor in MM. Further studies are underway to
evaluate the possible application of ADAMTS9 as a biomarker for the
diagnosis and treatment of MM and to explore its possible impact on
the pathways involved in MM development.
Acknowledgements
The authors thank Dr Jian Hou (The Second Military
Medical University, Shanghai, China) for providing the MM cells and
the Laboratory of Hematology (the First Affiliated Hospital of
Chongqing Medical University) for providing BM samples. The present
study was supported by grants from the National Natural Science
Foundation of China (nos. 31171243 and 81072148).
References
|
1
|
Hatzimichael E, Dasoula A, Benetatos L, et
al: Study of specific genetic and epigenetic variables in multiple
myeloma. Leuk Lymphoma. 51:2270–2274. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rajumar SV and Greipp PR: Prognostic
factors in multiple myeloma. Hematol Oncol Clin North Am.
13:1295–1314. 1999. View Article : Google Scholar
|
|
3
|
Kyle RA and Rajkumar SV: Multiple myeloma.
N Engl J Med. 351:1860–1873. 2004. View Article : Google Scholar
|
|
4
|
Klein U, Jauch A, Hielscher T, et al:
Chromosomal aberrations +1q21 and del(17p13) predict survival in
patients with recurrent multiple myeloma treated with lenalidomide
and dexamethasone. Cancer. 117:2136–2144. 2011.
|
|
5
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar
|
|
6
|
Jost E, Gezer D, Wilop S, et al:
Epigenetic dysregulation of secreted Frizzled-related proteins in
multiple myeloma. Cancer Lett. 281:24–31. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boultwood J and Wainscoat JS: Gene
silencing by DNA methylation in haematological malignancies. Br J
Haematol. 138:3–11. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuregir OO, Yurtcu E, Kizilkilic E, Kocer
NE, Ozdogu H and Sahin FI: Detecting methylation patterns of p16,
MGMT, DAPK and E-cadherin genes in multiple myeloma patients. Int J
Lab Hematol. 32:142–149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Braggio E, Maiolino A, Gouveia ME, et al:
Methylation status of nine tumor suppressor genes in multiple
myeloma. Int J Hematol. 91:87–96. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ullmannova-Benson V, Guan M, Zhou X, et
al: DLC1 tumor suppressor gene inhibits migration and invasion of
multiple myeloma cells through RhoA GTPase pathway. Leukemia.
23:383–390. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galm O, Yoshikawa H, Esteller M, Osieka R
and Herman JG: SOCS-1, a negative regulator of cytokine signaling,
is frequently silenced by methylation in multiple myeloma. Blood.
101:2784–2788. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Levy GG, Nichols WC, Lian EC, et al:
Mutations in a member of the ADAMTS gene family cause thrombotic
thrombocytopenic purpura. Nature. 413:488–494. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lo PH, Lung H, Cheung AK, et al:
Extracellular protease ADAMTS9 suppresses esophageal and
nasopharyngeal carcinoma tumor formation by inhibiting
angiogenesis. Cancer Res. 70:5567–5576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lung HL, Lo PH, Xie D, et al:
Characterization of a novel epigenetically-silenced,
growth-suppressive gene, ADAMTS9 and its association with lymph
node metastases in nasopharyngeal carcinoma. Int J Cancer.
123:401–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang C, Shao Y, Zhang W, et al:
High-resolution melting analysis of ADAMTS9 methylation levels in
gastric, colorectal and pancreatic cancers. Cancer Gene Cytogenet.
196:38–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuo HK, Griffith JD and Kreuzer KN:
5-Azacytidine induced methyltransferase-DNA adducts block DNA
replication in vivo. Cancer Res. 17:8248–8254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sidransky D: Emerging molecular markers of
cancer. Nat Rev Cancer. 2:210–219. 2002. View Article : Google Scholar
|
|
18
|
de Carvalho F, Colleoni GW, Almeida MS,
Carvalho AL and Vettore AL: TGFβR2 aberrant methylation is a
potential prognostic marker and therapeutic target in multiple
myeloma. Int J Cancer. 125:1985–1991. 2009.
|
|
19
|
Lo PH, Leung AC, Kwok CY, et al:
Identification of a tumor suppressive critical region mapping to
3p14.2 in esophageal squamous cell carcinoma and studies of a
candidate tumor suppressor gene, ADAMTS9. Oncogene. 26:148–157.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Z, Zhang W, Shao Y, et al:
High-resolution melting analysis of ADAMTS18 methylation levels in
gastric, colorectal and pancreatic cancers. Med Oncol. 27:998–1004.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moncada-Pazos A, Obaya AJ, Fraga MF, et
al: The ADAMTS12 metalloprotease gene is epigenetically silenced in
tumor cells and transcriptionally activated in the stroma during
progression of colon cancer. J Cell Sci. 122:2906–2913. 2009.
View Article : Google Scholar
|
|
22
|
Viloria CG, Obaya AJ, Moncada-Pazos A, et
al: Genetic inactivation of ADAMTS15 metalloprotease in human
colorectal cancer. Cancer Res. 69:4926–4934. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dunn JR, Panutsopulo D, Shaw MW, et al:
METH-2 silencing and promoter hypermethylation in NSCLC. Br J
Cancer. 91:1149–1154. 2004.PubMed/NCBI
|
|
24
|
Dubail J, Kesteloot F, Deroanne C, et al:
ADAMTS-2 functions as anti-angiogenic and anti-tumoral molecule
independently of its catalytic activity. Cell Mol Life Sci.
67:4213–4232. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
EI Hour M, Moncada-Pazos A, Blacher S, et
al: Higher sensitivity of Adamts12-deficient mice to tumor growth
and angiogenesis. Oncogene. 29:3025–3032. 2010.PubMed/NCBI
|
|
26
|
Dunn JR, Reed JE, du Plessis DG, et al:
Expression of ADAMTS-8, a secreted protease with antiangiogenic
properties, is downregulated in brain tumours. Br J Cancer.
94:1186–1193. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Koo BH, Coe DM, Dixon LJ, et al: ADAMTS9
is a cell-autonomously acting, anti-angiogenic metalloprotease
expressed by microvascular endothelial cells. Am J Pathol.
176:1494–1504. 2010. View Article : Google Scholar : PubMed/NCBI
|