Introduction
Ischemic stroke is a worldwide public health issue.
Ischemic stroke causes brain dysfunction and is one of the most
frequent causes of mortality. The most effective strategy for
treating the injury and limiting infarct size is the early
restoration of coronary blood flow to the ischemic myocardium.
However, this treatment is often associated with functional and
structural damage during reperfusion (1), which leads to cerebral edema, brain
hemorrhage and even neuronal death. Among the animal models, middle
cerebral artery occlusion (MCAO) followed by reperfusion is most
frequently used as the animal model of focal cerebral ischemia and
resembles human ischemic stroke (2). The molecular mechanism of ischemic
brain injury is not completely understood. Mitochondria have been
demonstrated to be involved in the regulation of the apoptosis
process and are important for ischemic cell death. Mitochondria are
the source of energy for sustaining life and in addition to their
role as energy-producing organelles, they participate in the
majority of physiological processes, including the cell cycle, the
production of reactive oxygen species (ROS), apoptosis and ion
balance (3,4). Significant progress has been made in
previous years to understand the mechanisms of mitochondrial
biology.
Mitochondrial channels, whose activities are linked
to a large number of mitochondrial functions and pathologies, have
received significant attention (5). Over 50 years ago, using a variety of
techniques under experimental conditions, the mitochondrial calcium
uniporter (MCU) was identified to be responsible for the uptake of
Ca2+ by mitochondria, driven by the large electrical
potential difference between the cytosol and the mitochondrial
matrix. Previous studies have demonstrated that under pathological
conditions, including ischemia/reperfusion (I/R) injury,
mitochondria accumulate significant amounts of Ca2+ from
the cytosol via MCU. Increases in mitochondrial Ca2+
concentration ([Ca2+]m) induce the opening of
the mitochondrial permeability transition pore (MPTP) leading to
inhibition of ATP synthesis, increased ROS production, cytochrome
c release and cell death by apoptosis (6,7).
Ruthenium red (RR) and Ru360 block the MCU to reduce the
Ca2+ influx and, therefore, exert a beneficial effect
during I/R injury by preventing Ca2+ accumulation
(8).
Oxidative phosphorylation, involving the electron
transport chain (ETC) and ATP synthase, provides the vast majority
of cellular energy and drives all cellular processes. The ETC is
composed of NADH dehydrogenase (complex I), succinate dehydrogenase
(complex II), cytochrome c reductase (complex III) and
cytochrome c oxidase (complex IV). Previous studies have
revealed that under pathological conditions, including I/R injury,
the ETC causes cell damage and even triggers death processes due to
depressed ATP production and the generation of ROS (9). ROS are produced via the ETC, however,
these chemically reactive molecules also damage electron transport
complexes (10), leading to
further respiratory dysfunction and increases in ROS production,
which results in a positive feedback cycle during I/R. Appropriate
treatment strategies may be administered to break this cycle and
protect mitochondrial structure and function.
Our previous studies have revealed that ROS
production is reduced by inhibiting MCU (11). Thus, the current study was based on
the hypothesis that electron transport complexes and cerebral
infarction area are affected by the activity of the MCU in
ischemic/reperfused rats. ATP levels, ROS production, changes in
mitochondrial membrane potential (ΔΨm) and HE and TUNEL
staining results were analyzed to to determine the mechanisms
underlying the effects of MCU activity in a rat model of cerebral
I/R injury.
Materials and methods
Chemicals and reagents
RR, spermine and rhodamine 123 were purchased from
Sigma-Aldrich (St Louis, MO, USA). The ATP assay kit was purchased
from Beyotime Institute of Biotechnology (Jiangsu, China).
Mitochondria isolation and assay kits for mitochondrial complex
studies were purchased from Genmed Scientifics Inc. (Shanghai,
China). The Bradford protein assay kit was purchased from Nanjing
Jiancheng Bioengineering Institute (Nanjing, China).
Animals
Male Wistar rats weighing 250–300 g (supplied by the
experimental animal center of Qingdao Drug Inspection Institute,
Qingdao, China) were randomly divided into 4 groups of 12 animals:
I (sham), without coronary artery ligation, underwent identical
surgical procedures as the I/R group; II (I/R), received saline
solution (0.9%) 30 min prior to MCAO, i.e., ischemia was induced
for 2 h followed by 24 h of reperfusion; III (I/R + RR), received a
bolus injection of RR dissolved in saline solution 30 min prior to
MCAO; IV (I/R + Sper), received a bolus injection of spermine
dissolved in saline solution 30 min prior to MCAO.
MCAO model
MCAO rats were generated as described previously
(12). Briefly, rats were
anesthetized with 10% chloral hydrate (350 mg/kg, i.p.). The right
common carotid artery (CCA), internal carotid artery (ICA) and
external carotid artery (ECA) were exposed via a midline incision
on the neck. Next, the CCA and ECA were ligated (near the
bifurcation) with 4-0 surgical sutures and the ICA was clipped with
an artery clip. After a small incision was made in the CCA, a nylon
filament with a diameter of 0.285 mm was introduced into the ICA
(18–20 mm from the external-internal carotid artery bifurcation)
through the CCA. The nylon filament was maintained in position for
2 h and was then carefully removed to restore the blood flow.
During surgery, body temperature was maintained at 36.5–37.5°C
using a heating pad.
Neurological deficit scoring
evaluation
Neurological deficits were evaluated following 2 h
ischemia and 24 h reperfusion according a scale system described
previously (13): 0, normal, no
neurological deficit signs; 1, failure to extend contralateral
forepaw on lifting of the animal by the tail; 2, circling to the
contralateral side, but normal posture at rest; 3, falling to the
contralateral side at rest; 4, no spontaneous locomotor activity.
The model was confirmed at scores 2–4.
Measurement of infarct volumes
To analyze the infarct volumes, TTC
(2,3,5-triphenyltetrazolium chloride) staining was used. Rats were
sacrificed 24 h following reperfusion, brains were removed rapidly
and frozen at −20°C for 5 min. The brains were sectioned into 2-mm
thick coronal slices using a brain-sectioning block and stained
with standard 1% TTC for 15 min at 37°C. Images of sections were
captured and analyzed using the Image-Pro Plus 5.1 analysis system
(Media Cybernetics, Rockville, MD, USA). Infarct volume (%HLV) was
calculated using the following equation: %HLV = {[total infarct
volume − (right hemisphere volume − left hemisphere volume)]/left
hemisphere volume} × 100.
HE staining
Following reperfusion (24 h), rats were anesthetized
and perfused with 200 ml 0.9% sodium chloride, followed by 100 ml
4% paraformaldehyde (PFA) in 0.1 M phosphate-buffered saline (PBS,
pH 7.4) through the left ventricle. Brains were removed and fixed
in 4% PFA overnight. Sections (3-mm thick) cut coronally from optic
chiasma to occipital pole were embedded in paraffin and cut into
several segments (4-μm thick). Coronal sections were stained with
hematoxylin-eosin. Sections were also used for TUNEL staining.
TUNEL staining
An in situ cell death detection kit (Roche
Diagnostics GmbH, Mannheim, Germany) was used to detect apoptotic
cell death. According to the manufacturer’s instructions with minor
modifications, paraffin embedded sections were deparaffinized and
rehydrated by graded ethanol, followed by treatment with proteinase
K for 15 min and 3% H2O2 for 10 min at room
temperature. Following three 10 min washes in PBS, sections were
incubated with terminal deoxynucleotidyl transferase at 37°C for 2
h. Sections were washed in PBS three times for 10 min each and
further incubated with anti-digoxigenin conjugate for 30 min at
37°C. Following washing, 3,3′-diaminobenzidine was used to
visualize apoptotic cells. Images were captured at a magnification
of ×400 using the Nikon ECLIPSE TE300 fluorescence microscope
(Nikon Instruments, Inc., Melville, NY, USA).
Isolation of mitochondria
Brain mitochondria were isolated by differential
centrifugation, using the Functional Mitochondria Isolation kit
(Genmed Scientifics, Inc., Wilmington, DE, USA). All procedures
were performed on ice to maintain the temperature at 4°C. Rats were
anesthetized, decapitated and the perifocal penumbra zone of the
brain was immediately removed. Tissue was immediately placed in
ice-cold PBS to remove impurities, then processed into fine pieces.
The tissue was homogenized in isolation medium using a glass
homogenizer. The homogenate was centrifuged at 1,000 × g for 10
min. The supernatant was retained and centrifuged at 10,000 × g for
10 min. The resulting pellet was resuspended and rinsed by
centrifuging at 10,000 × g for 5 min. The final mitochondrial
pellet was resuspended in reaction buffer and stored at −80°C.
Protein concentration was determined with the Bradford protein
assay using BSA as a standard (14).
Measurement of mitochondrial complex
activity
Mitochondrial freeze-thaw cycles were repeated 3
times to enable enzyme release. The activities of mitochondrial
complexes I–IV were measured using an assay kit according to the
manufacturer’s instructions and a microplate reader (Tecan Group
Ltd., Männedorf, Switzerland).
Measurements of intracellular ATP
levels
ATP levels were determined using the ATP detection
kit (Beyotime Institute of Biotechnology) following the
luciferin-luciferase method (15).
Briefly, tissue was homogenized with a glass homogenizer in a lysis
buffer from the ATP detection kit, followed by centrifuging at
12,000 × g for 5 min at 4°C. The supernatant was retained for the
ATP test. Supernatant or standard buffer (both 100 μl) were mixed
with 100 μl ATP detection working dilution on a black plate and
luminescence was measured immediately. Standard curves were
generated, to which the ATP level was referred and the protein
concentration of each treatment group was determined using the
Bradford protein assay.
Measurement of mitochondrial membrane
potential
Flow cytometry was used to monitor ΔΨm
according to the method described previously (16) with minor modifications. Rhodamine
123, a fluorescent cationic dye, uses a transmembrane
potential-dependent mechanism to enter the mitochondria. The tissue
was digested with pancreatic enzymes and made into suspended cells,
which were washed with PBS. In the dark, cells were loaded with
Rhodamine 123 (final concentration, 5 μmol/l) at 37°C for 30 min
(17) and then washed twice with
PBS. Following incubation, fluorescence was determined using flow
cytometry (FC500 MPL; Beckman Coulter, Miami, FL, USA) at an
excitation wavelength of 488 nm and emission wavelength of 530 nm.
Data were analyzed using the CXP 2.1 software package.
Measurement of intracellular ROS
generation
2′,7′-Dihydrodichlorofluorescein diacetate
(DCFH-DA), a membrane-permeable probe, was used to evaluate ROS
generation (18). The
non-fluorescent dye freely diffuses through the cell membrane and
is hydrolyzed to the nonfluorescent DCFH by intracellular
esterases. When DCFH is oxidized by ROS, it yields the fluorescent
product, DCF. A sample was incubated with DCFH-DA (100 μM) in the
dark at 37°C for 20 min and DCF fluorescence intensity was measured
using a fluorescence spectrophotometer (Tecan Group Ltd.) at an
excitation wavelength of 488 nm and an emission wavelength of 525
nm.
Statistical analysis
SPSS 17.0 statistical software was used to perform
statistical analysis. P<0.05 was considered to indicate a
statistically significant difference. All data are expressed as
mean ± SD. ANOVA was used to compare the difference between
groups.
Results
Role of the MCU in neurological deficits
following I/R injury
Following reperfusion (24 h), rats in the sham group
exhibited no neurological deficits. Neurological deficit scores
were observed to be significantly higher in the I/R and I/R + Sper
groups than in the sham group (P<0.01), however, no significant
differences were observed between the I/R and I/R + Sper groups. By
contrast, scores were significantly lower in the I/R + RR group
(P<0.01) than in the I/R group (Fig. 1).
Role of the MCU in cerebral infarct
volume
Infarct volume measurements revealed that rats from
the I/R group exhibited a larger infarct area than the rats from
the sham group, in which no infarction was observed. In the I/R
group, irregular pale areas supplied by the middle cerebral artery
were observed in sections. Treatment with RR (2.5 mg/kg) in the I/R
+ RR group decreased the %HLV from that in the I/R group
(26.00±1.71 vs. 35.43±0.74%, respectively; P<0.01). Spermine (5
mg/kg) in the I/R + Sper group increased the %HLV (36.57±1.31%),
however, the change in infarct volume from that in brains that
underwent I/R only was not determined to be significant (Fig. 2).
Role of the MCU in neuronal damage and
cell apoptosis of brain tissue
HE staining was used to evaluate the
histopathological values at 24 h following reperfusion. No brain
infarction was found in the sham group and the cell outline was
clear, the structure was compact and the nucleolus was clearly
visible. Cells in the I/R and I/R + Sper groups were arranged
sparsely and revealed pyknotic nuclei. In addition, cell outlines
were undefined, structures were disordered and deformation of cells
was severe. In the I/R + RR group, there were fewer necrotic cells,
cell outlines were relatively clear and cell structures were
compact (Fig. 3A).
 | Figure 3Effect of mitochondrial calcium
uniporter activity on neuronal damage and cell apoptosis. (A) HE
and TUNEL staining for neurons in sham, I/R, I/R + RR and I/R +
Sper groups were assessed at 24 h following reperfusion. (B) The
proportion of TUNEL-positive cells increased markedly in the I/R
group compared with that in the sham group. RR treatment
significantly reduced the number of TUNEL-positive cells compared
with that in the I/R group. Following treatment with spermine, the
number of TUNEL-positive cells increased significantly.
*P<0.01, vs. sham; △P<0.01, vs. I/R;
#P<0.01, vs. I/R + Sper. Each group, n=3. I/R,
ischemia/reperfusion; RR, ruthenium red; Sper, spermine. |
The TUNEL assay was used to determine cell
apoptosis. TUNEL-positive cells with apoptotic bodies and dark
staining were considered to be apoptotic cells (Fig. 3A). Apoptotic cells were almost
unobservable in the sham group (6.50±0.85%). The numbers of
TUNEL-positive cells in the I/R and I/R + Sper groups were higher
than those in the sham rats (P<0.01) and were slightly higher in
the I/R + Sper group than in the I/R group. The number of
TUNEL-positive cells was significantly lower in the I/R + RR group
(63.63±1.60%) than in the I/R group, indicating that RR treatment
significantly ameliorated cell survival and inhibited apoptosis
(Fig. 3B).
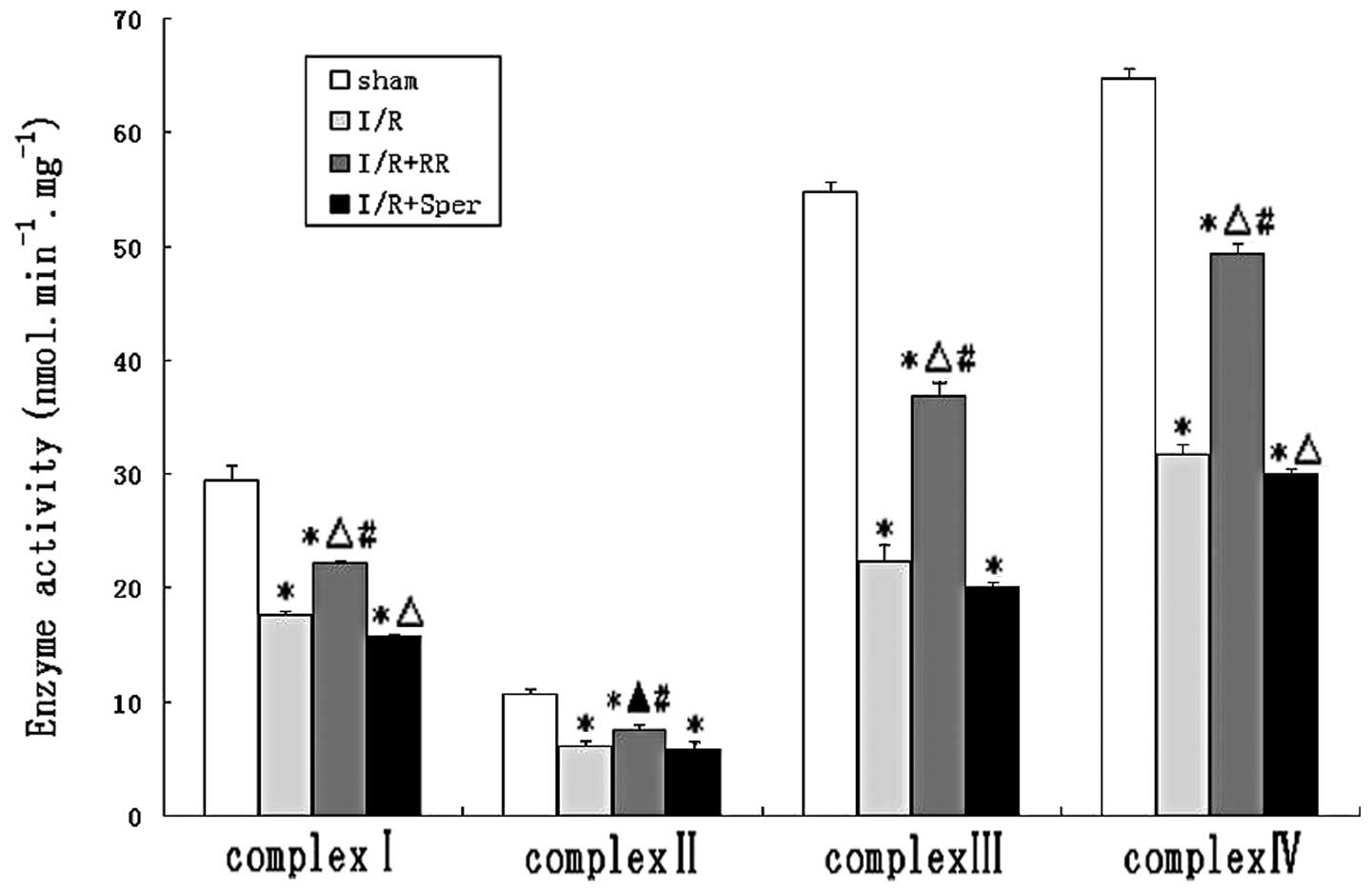
Effect of MCU on mitochondrial complex
activities
To evaluate the effect of MCU activity on
mitochondrial energy metabolism, the enzyme activities of complexes
I–IV were measured in mitochondria isolated from brains. Activities
of complexes I, II, III and IV were demonstrated to be decreased by
~40, 43, 59 and 51%, respectively, in animals suffering from I/R
and increased by ~15, 12, 26 and 27%, respectively, following RR
treatment. The activities of the ETC complexes were lower in the
I/R group than in the sham group (P<0.01). The results indicate
that I/R led to inhibition of mitochondrial complexes. RR treatment
significantly ameliorated the activities of complexes I, III and IV
(P<0.01) and complex II (P<0.05), while complex I, II, III
and IV activities decreased in the I/R + Sper group (Fig. 4).
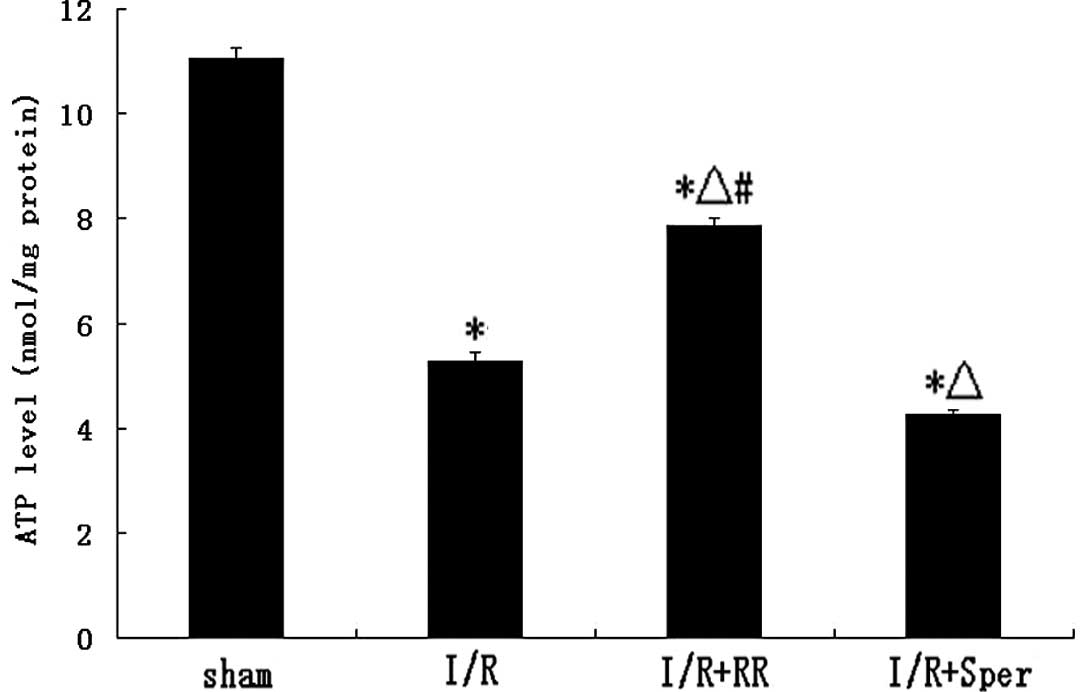
Effect of MCU on ATP levels
Fig. 5 demonstrates
that ATP levels in I/R rats decreased following 24 h reperfusion.
ATP levels in the RR-treated group were ~51% higher than in the I/R
group (P<0.01), whereas, following treatment with spermine, ATP
levels were lower than in the I/R group (P<0.01; Fig. 5).
Effect of MCU on mitochondrial membrane
potential
The mitochondrial membrane potential has been widely
used to study mitochondrial health. Rhodamine 123, a
mitochondrion-selective fluorescent dye which is rapidly
sequestered by mitochondria, was used as a marker of membrane
disruption. The X-mean was found to be significantly lower in I/R
cells (5.67±0.32; P<0.01) than in those of the sham group
(Fig. 6B), with the wave moving
left (Fig. 6A), indicative of a
significant loss of membrane potential. RR treatment increased the
X-mean significantly (8.76±0.45; P<0.01), with the wave moving
right compared with the I/R group. However, the wave did not reach
normal levels (sham group). RR treatment attenuated the dissipation
of mitochondrial membrane potential caused by reperfusion injury.
In the presence of spermine to induce MCU opening, the X-mean
increase was higher than in the I/R group (4.80±0.30; P<0.05),
indicating mitochondrial depolarization (Fig. 6).
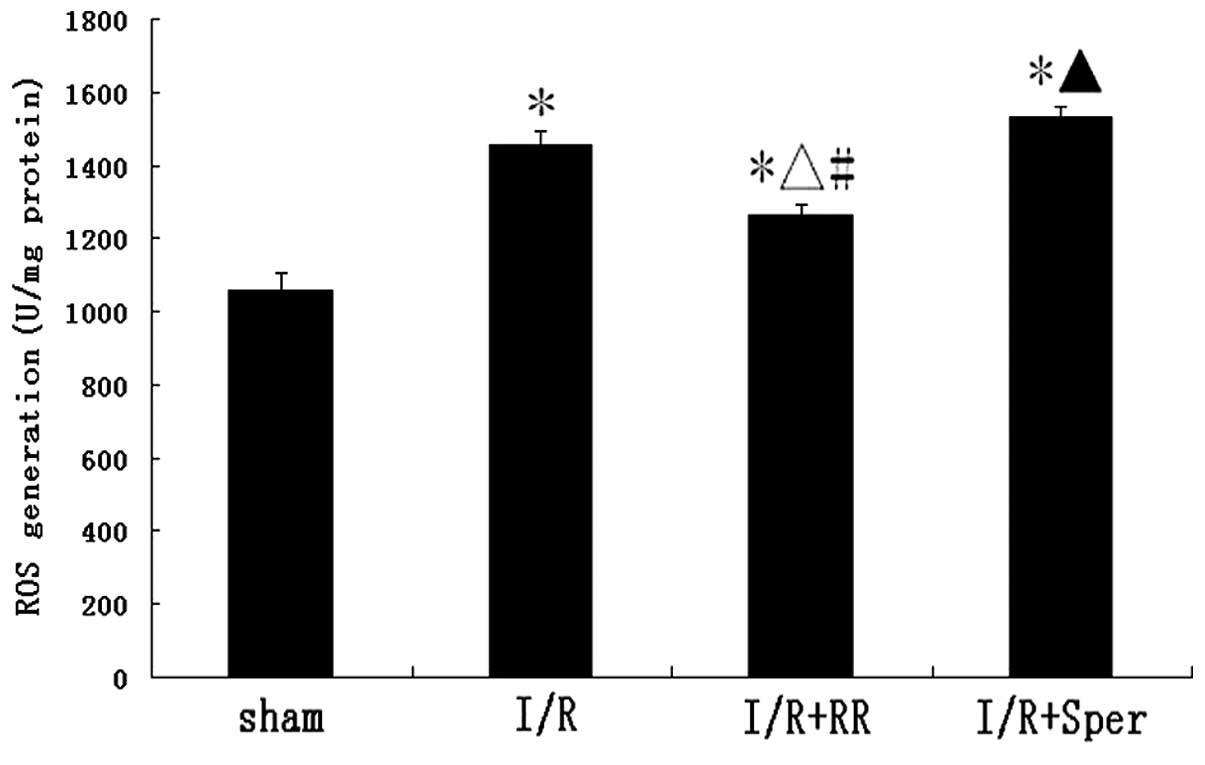
Effect of MCU activity on intracellular
ROS generation
To observe whether ROS generation is affected by MCU
activation, ROS generation was monitored by detection of DCFH-DA
fluorescence. In the sham group, low levels of DCFH-DA fluorescence
were observed. There was a marked increase of fluorescence in the
I/R group, indicative of an increase in ROS production. Following
treatment with RR, ROS generation was observed to decrease
significantly (P<0.01). By contrast, as revealed in Fig. 7, fluorescence in the I/R + Sper
group increased markedly and was higher than that in the I/R group
(P<0.05). In the present study, MCU activity was demonstrated to
regulate intracellular ROS generation.
Discussion
Ischemic stroke is one of the leading causes of
mortality and disability in the world. However, at present, only a
limited number of therapeutic strategies have been found to target
ischemic brain injury and it remains a major public health issue.
MCAO followed by reperfusion has been widely used to study ischemia
mechanisms and potential interventions. In the present study, MCU
inhibition was found to have anti-apoptotic and neuroprotective
effects during cerebral I/R injury in rats. The results revealed
that treatment with RR, which blocks MCU, significantly ameliorated
neurological deficit scores and led to a significant reduction in
cerebral infarction during MCAO. HE staining revealed that RR
significantly reduced neuronal injury. In addition, RR was observed
to exhibit neuroprotective effects against ischemia-induced
apoptosis of neuronal cells. RR also demonstrated protective
effects against reperfusion injury in the heart (19,20).
By contrast, treatment with spermine, an activator of the
uniporter, led to converse results. MCU was demonstrated to have a
regulatory effect in focal cerebral I/R injury, consistent with
previous studies (21–23). In addition, the mechanism by which
MCU regulates cerebral I/R injury was hypothesized to be associated
with improved mitochondrial energy metabolism due to MCU
inhibition.
Mitochondria play a crucial role in the production
of energy and are the site of the majority of ATP generation.
Decreases in cellular ATP levels, impaired mitochondrial oxidative
respiration and large influxes of Ca2+ that result in
cellular excitotoxicity and apoptosis (24) are present during I/R. The
inhibition of key mitochondrial respiratory complexes has been
observed to be the cause of ischemia-induced mitochondrial
dysfunction (25). Cells are
almost entirely dependent on mitochondrially generated ATP for
their energy and, therefore, a defect at any level of the
mitochondrial oxidative phosphorylation machinery has profound
effects on brain function. As a key event in I/R injury,
mitochondrial Ca2+ overload does not always result in
MPTP opening and cytochrome c release to detrimentally
affect mitochondrial function. Overload is known to lead to
inhibition of mitochondrial respiratory complexes, with subsequent
enhancement of ROS generation, which then results in the inhibition
of respiratory complexes and mitochondrial dysfunction.
The MCU, which has been investigated for ~40 years,
is a highly selective ion channel located in the mitochondrial
inner membrane. It is responsible for the uptake of Ca2+
in mitochondria driven by the large electrical potential difference
between the cytosol and mitochondrial matrix (26). Ca2+ transport activity
is inhibited by RR and its associated compound, RuR360 and is also
modulated by aliphatic polyamines, including spermine and
aminoglycosides. Although MCU has recieved considerable attention
for decades, this gated channel has not been cloned or isolated and
its molecular identity remains controversial. Uncoupling protein
(UCP) 2 and UCP3 were previously reported to be the essential
components of the uniporter machinery (27), however, the results remain
controversial and further studies are required to definitively
elucidate the molecular identity of the channels. In a previous
study, a 54 kDa protein known as mitochondrial calcium uptake 1 was
identified, whose silencing regulates mitochondrial Ca2+
uptake (28). As a single-pass
transmembrane protein, it is unlikely to function as a
Ca2+ channel. Instead, it is likely to act as a
fundamental subunit of the complex for the uptake machinery. In
2011, MCU was identified by analyzing 14 genes in detail and
concluded that the protein Ccdc109A may be a component of MCU
(29,30). A subsequent study by these authors
hypothesized that MCU is an inner mitochondrial membrane protein
with the C-terminus facing the intermembrane space (29,30).
Mitochondrial Ca2+is key to the
regulation of mitochondrial functions, ranging from mediating
signaling pathways between the cytosol and the mitochondrial matrix
to modulating mitochondrial energy metabolism. However, the
molecular mechanisms underlying mitochondrial Ca2+
transport remain unclear (31).
Within the mitochondrial matrix, enhanced Ca2+ levels
improve electron transport and increase NADH and ATP production
(32). Therefore, mitochondrial
ATP output is altered to meet cellular ATP demands and, under
specific conditions, reduce ROS formation (33). However, under pathological
conditions, including I/R, Ca2+ accumulation in
mitochondria has been proposed to be significant in cellular injury
(34). ROS increase the cytosolic
Ca2+ concentration, affecting the sarcoplasmic reticulum
and sarcolemmal membranes during I/R (35), and then increases
[Ca2+]m(20). The overload in mitochondrial
Ca2+, together with cyclophilin D, induce MPTP opening
(36), which releases cytochrome
c, Smac/DIABLO and apoptosis-inducing factors involved in
apoptotic death signaling (37).
In addition, mitochondrial Ca2+ overload triggers the
generation of factors, including ROS and free fatty acids (38), and also promotes MPTP opening.
There are several mechanisms that are hypothesized to be involved
in Ca2+-induced ROS production. Mitochondrial complexes
are known to be damaged when animals are subjected to I/R, causing
an excessive release of ROS. Under physiological conditions,
Ca2+ may be a partial inhibitor of the ETC, leading to
ROS production. Respiratory chain enzymes, NADH dehydrogenase
(complex I) and cytochrome c oxidase (complex IV),
participate in energy metabolism and the generation of ROS and are
vulnerable to ROS attack. Intracellular ATP production is reported
to be the prime target of free radicals in hypoxia/ischemia
(39). NADH dehydrogenase and
cytochrome c oxidase activities were observed to decrease following
I/R, which may be the direct or indirect result of ROS damage in
the penumbra zone. Treatment with RR has been reported to protect
against the loss of complex I activity in isolated cardiomyocytes
subjected to hypoxia/reoxygenation (40), therefore, it has been hypothesized
that mitochondrial Ca2+ uptake during reoxygenation is
involved in the mechanism. A number of previous studies have
reported that inhibition of complex I, in combination with
Ca2+ overload, leads to enhanced ROS generation in in
vitro and in vivo systems (41,42).
Under physiological conditions, Ca2+ also dissociates
cytochrome c from the mitochondrial inner membrane by
triggering MPTP opening and cytochrome c blocks the
respiratory chain at complex III effectively, causing an increase
in ROS production (43,44). The spectra of cytochromes
a/a3 is reported to be altered by Ca2+ in
isolated complex IV (45).
Furthermore, Ca2+ stimulation of the tricarboxylic acid
(TCA) cycle increases the metabolic rate, which results in
increased respiratory chain electron leakage. Ca2+
stimulation of nitric oxide synthase (NOS), which generates NO,
inhibits the respiratory chain at complex IV (46) and subsequently enhances ROS
generation from the Q cycle. NO demonstrates a marked contribution
to changes in mitochondrial energy metabolism during the I/R
transition. In addition, NO and Ca2+ are known to
inhibit complex I together and may also increase ROS generation by
this complex (47,48).
In this study, ROS production was observed to be
increased in animals who had undergone I/R, but was attenuated by
the MCU inhibitor, RR, which permeates slowly into the cell and
specifically inhibits mitochondrial Ca2+ uptake. This
observation is consistent with a previous study which reported that
RR decreased ROS production, induced by Ca2+ overload
(47). Ru360, a specific MCU
inhibitor, was previously identified to be important in the
modulation of ROS under conditions involving excessive
mitochondrial Ca2+ overload (49). In the present study, activation of
the uniporter with spermine (5 mg/kg) caused the levels of ROS
production to increase, whereas RR caused them to decrease,
indicating that MCU activities are important for the regulation of
ROS production during brain I/R injury. In addition, analysis of
the respiratory chain demonstrated an improvement in I/R-related
mitochondrial respiratory complex I, II, III and IV dysfunction
with RR treatment, with the opposite results following spermine
treatment. However, the underlying mechanisms remain to be
elucidated. MCU has been previously hypothesized to affect
mitochondrial respiratory complexes via regulation of mitochondrial
Ca2+ and/or ROS. In addition, RR protection of
mitochondrial respiratory complexes I–IV may increase ATP
production, which is extremely important for the preservation of
ATP-dependent cellular processes in I/R. These hypotheses are
consistent with results of the present study. ΔΨm is
known to be a pivotal factor for the determination of cellular
survival during I/R. It has previously been reported that
ΔΨm hyperpolarization is associated with the generation
of ROS which, at specific levels, may trigger mitochondrial
membrane depolarization (50). A
previous study reported that ΔΨm decrease was an
initiator and was also a consequence of MPTP opening (51). As as an inhibitor of MCU, Ru360 may
normalize mitochondrial membrane depolarization by preventing the
opening of the MPTP during I/R (22). The results of the present study
revealed changes in ΔΨm, specifically, membrane
depolarization during I/R, normalization by RR and accelerated
collapse following spermine treatment, that are indicative of the
regulatory functions of MCU on ΔΨm during I/R.
In conclusion, the results of the present study
indicate that MCU activities are important in I/R by regulating
mitochondrial energy metabolism. Blocking the uniporter with RR
increases the functional recovery of brains subjected to I/R and
the mechanism may be associated with improved energy metabolism.
These results are likely to contribute to the understanding of MCU
in I/R, however, additional studies must be performed to further
elucidate the mechanism. In addition, the identification of novel
effective drugs targeting the MCU for use in reperfusion therapy is
likely to be of significant interest.
References
|
1
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
2
|
Boyko M, Ohayon S, Goldsmith T, et al:
Cell-free DNA - a marker to predict ischemic brain damage in a rat
stroke experimental model. J Neurosurg Anesthesiol. 23:222–228.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnson DT, Harris RA, Blair PV and
Balaban RS: Functional consequences of mitochondrial proteome
heterogeneity. Am J Physiol Cell Physiol. 292:C698–C707. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: more than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun Y, Deng T, Lu N, Yan M and Zheng X:
B-type natriuretic peptide protect cardiomyocytes at reperfusion
via mitochondrial calcium uniporter. Biomed Pharmacother.
64:170–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sullivan PG, Rabchevsky AG, Waldmeier PC
and Springer JE: Mitochondrial permeability transition in CNS
trauma: cause or effect of neuronal cell death? J Neurosci Res.
79:231–239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Demaurex N and Distelhorst C: Cell
biology. Apoptosis - the calcium connection. Science. 300:65–67.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan Y, Liu J, Wei C, et al: Bidirectional
regulation of Ca2+ sparks by mitochondria-derived
reactive oxygen species in cardiac myocytes. Cardiovasc Res.
77:432–441. 2008.
|
|
9
|
Young TA, Cunningham CC and Bailey SM:
Reactive oxygen species production by the mitochondrial respiratory
chain in isolated rat hepatocytes and liver mitochondria studies
using myxothiazol. Arch Biochem Biophys. 405:65–72. 2002.
View Article : Google Scholar
|
|
10
|
Chen Q, Moghaddas S, Hoppel CL and
Lesnefsky EJ: Ischemic defects in the electron transport chain
increase the production of reactive oxygen species from isolated
rat heart mitochondria. Am J Physiol Cell Physiol. 294:C460–C466.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu N, Wang S, Wang P, et al: The calcium
uniporter regulates the permeability transition pore in isolated
cortical mitochondria. Neural Regen Res. 72:109–113.
2012.PubMed/NCBI
|
|
12
|
Yang JP, Liu XF, Liu HJ, Xu GL and Ma YP:
Extracellular signal-regulated kinase involved in NGF/VEGF-induced
neuroprotective effect. Neurosci Lett. 434:212–217. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu X, Chua KW, Chua CC, et al: Synergistic
protective effects of humanin and necrostatin-1 on hypoxia and
ischemia/reperfusion injury. Brain Res. 1355:189–194. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
St John JB: Determination of ATP in
Chlorella with the luciferin-luciferase enzyme system. Anal
Biochem. 37:409–416. 1970.
|
|
16
|
Vander Heiden MG, Chandel NS, Williamson
EK, et al: Bcl-xL regulates the membrane potential and volume
homeostasis of mitochondria. Cell. 91:627–637. 1997.PubMed/NCBI
|
|
17
|
Emaus RK, Grunwald R and Lemasters JJ:
Rhodamine 123 as a probe of transmembrane potential in isolated
rat-liver mitochondria: spectral and metabolic properties. Biochim
Biophys Acta. 850:436–448. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
LeBel CP, Ischiropoulos H and Bondy SC:
Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of
reactive oxygen species formation and oxidative stress. Chem Res
Toxicol. 5:227–231. 1992.
|
|
19
|
Ferrari R, di Lisa F, Raddino R and
Visioli O: The effects of ruthenium red on mitochondrial function
during post-ischaemic reperfusion. J Mol Cell Cardiol. 14:737–740.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miyamae M, Camacho SA, Weiner MVV and
Figueredo VM: Attenuation of postischaemic reperfusion injury is
related to prevention of [Ca2+]m overload in
rat hearts. Am J Physiol. 271:H2145–H2153. 1996.
|
|
21
|
Griffiths EJ: Mitochondrial calcium
transport in the heart: Physiological and pathological roles. J Mol
Cell Cardiol. 46:789–803. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
de García-Rivas GJ, Carvajal K, Correa F
and Zazueta C: Ru360, a specific mitochondrial calcium uptake
inhibitor, improves cardiac post-ischaemic functional recovery in
rats in vivo. Br J Pharmacol. 149:829–837. 2006.PubMed/NCBI
|
|
23
|
Clements-Jewery H: Mitochondria, the
calcium uniporter and reperfusion-induced ventricular fibrillation.
Br J Pharmacol. 149:811–813. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rashidian J, Iyirhiaro GO and Park DS:
Cell cycle machinery and stroke. Biochim Biophys Acta.
1772:484–493. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Almeida A, Allen KL, Bates TE and Clark
JB: Effect of reperfusion following cerebral ischaemia on the
activity of the mitochondrial respiratory chain in the gerbil
brain. J Neurochem. 65:1698–1703. 1995. View Article : Google Scholar
|
|
26
|
Bernardi P: Mitochondrial transport of
cations: channels, exchangers and permeability transition. Physiol
Rev. 79:1127–1155. 1999.PubMed/NCBI
|
|
27
|
Trenker M, Malli R, Fertschai I,
Levak-Frank S and Graier WF: Uncoupling proteins 2 and 3 are
fundamental for mitochondrial Ca2+ uniport. Nat Cell
Biol. 9:445–452. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Perocchi F, Gohil VM, Girgis HS, et al:
MICU1 encodes a mitochondrial EF hand protein required for
Ca2+ uptake. Nature. 467:291–296. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
De Stefani D, Raffaello A, Teardo E, Szabo
I and Rizzuto R: A forty-kilodalton protein of the inner membrane
is the mitochondrial calcium uniporter. Nature. 476:336–340.
2011.PubMed/NCBI
|
|
30
|
Baughman JM, Perocchi F, Girgis HS, et al:
Integrative genomics identifies MCU as an essential component of
the mitochondrial calcium uniporter. Nature. 476:341–345. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dash RK and Beard DA: Analysis of cardiac
mitochondrial Na+/Ca2+ exchanger kinetics
with a biophysical model of mitochondrial Ca2+ handling
suggests a 3:1 stoichiometry. J Physiol. 586:3267–3285.
2008.PubMed/NCBI
|
|
32
|
Jouaville LS, Pinton P, Bastianutto C,
Rutter GA and Rizzuto R: Regulation of mitochondrial ATP synthesis
by calcium: evidence for a long-term metabolic priming. Proc Natl
Acad Sci USA. 96:13807–13812. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Komary Z, Tretter L and Adam-Vizi V:
H2O2 generation is decrease by calcium in
isolated brain mitochondria. Biochim Biophys Acta. 1777:800–807.
2008.
|
|
34
|
Rizzuto R, Marchi S, Bonora M, et al:
Ca2+ transfer from the ER to mitochondria: when, how and
why. Biochim Biophys Acta. 1787:1342–1351. 2009.PubMed/NCBI
|
|
35
|
Krause SM, Jacobus WE and Becker LC:
Alterations in cardiac sarcoplasmic reticulum calcium transport in
the postischaemic ‘stunned’ myocardium. Circ Res. 65:526–530.
1989.
|
|
36
|
Basso E, Fante L, Fowlkes J, Petronilli V,
Forte MA and Bernardi P: Properties of the permeability transition
pore in mitochondria devoid of Cyclophilin D. J Biol Chem.
280:18558–18561. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Regula KM and Kirshenbaum LA: Apoptosis of
ventricular myocytes: a means to an end. J Mol Cell Cardiol.
38:3–13. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Starkov AA, Chinopoulos C and Fiskum G:
Mitochondrial calcium and oxidative stress as mediators of ischemic
brain injury. Cell Calcium. 36:257–264. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Halestrap A: Biochemistry: a pore way to
die. Nature. 434:578–579. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hardy L, Clark JB, Usmar VM, DR and Stone
D: Reoxygenation-dependent decrease in mitochondrial NADH: CoQ
reductase (complex I) activity in the hypoxic/reoxygenated rat
heart. Biochem J. 274:133–137. 1991.PubMed/NCBI
|
|
41
|
Sousa SC, Maciel EN, Vercesi AE and
Castilho RF: Ca2+-induced oxidative stress in brain
mitochondria treated with the respiratory chain inhibitor rotenone.
FEBS Lett. 543:179–183. 2003.
|
|
42
|
Yadava N and Nicholls DG: Spare
respiratory capacity rather than oxidative stress regulates
glutamate excitotoxicity after partial respiratory inhibition of
mitochondrial complex I with rotenone. J Neurosci. 27:7310–7317.
2007. View Article : Google Scholar
|
|
43
|
Grijalba M, Vercesi A and Schreier S:
Ca2+-induced increased lipid packing and domain
formation in submitochondrial particles. A possible early step in
the mechanism of Ca2+-stimulation generation of reactive
oxygen species by the respiratory chain. Biochemistry.
38:13279–13287. 1999.
|
|
44
|
Gincel D, Zaid H and Shoshan-Barmatz V:
Calcium binding and translocation by the voltage-dependent anion
channel: a possible regulatory mechanism in mitochondrial function.
Biochem J. 358:147–155. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wikström M and Saari H: A spectral shift
in cytochrome a induced by calcium ions. Biochim Biophys Acta.
408:170–179. 1975.PubMed/NCBI
|
|
46
|
Cleeter MW, Cooper JM, Darley-Usmar VM,
Moncada S and Schapira AH: Reversible inhibition of cytochrome c
oxidase, the terminal enzyme of the mitochondrial respiratory
chain, by nitric oxide. Implications for neurodegenerative
diseases. FEBS Lett. 345:50–54. 1994. View Article : Google Scholar
|
|
47
|
Votyakova TV and Reynolds IJ:
Ca2+-induced permeabilization promotes free radical
release from rat brain mitochondria with partially inhibited
complex I. J Neurochem. 93:526–537. 2005.
|
|
48
|
Vygodina TV, Dyuba AV and Konstantinov AA:
Effect of calcium ions on electron transfer between hemes a and
a(3) in cytochrome c oxidase. Biochemistry. 77:901–909.
2012.PubMed/NCBI
|
|
49
|
Tretter L, Biagioni Angeli E, Ardestani
MR, Goracci G and Adam-Vizi V: Reversible inhibition of hydrogen
peroxide elimination by calcium in brain mitochondria. J Neurosci
Res. 89:1965–1972. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ly JD, Grubb DR and Lawen A: The
mitochondrial membrane potential (ΔΨm) in apoptosis; an
update. Apoptosis. 8:115–128. 2003.
|