Introduction
Spinal cord injury (SCI) is a devastating
neurological injury that is associated with functional deficits and
is a major cause of disability. The initial tissue damage in SCI is
limited to the injury site. However, the damaged area gradually
expands over time. One of the primary causes of secondary damage is
toxicity caused by excessive excitatory amino acids released from
the injured tissue (1). Astrocytes
are involved in the reduction of excitotoxicity through the
conversion of glutamate into glutamine via glutamine synthetase
(GS) (2–4). However, it is not clear whether the
dysfunction of GS contributes to excitotoxicity-induced neuronal
loss in SCI.
The activation of astrocytes leads to the formation
of a glial scar, which inhibits axon regeneration and the
maturation of neuronal tissues (5). Our previous study found that GS
negatively regulates astrocyte migration and glial scar formation
in SCI, a process that is mediated by integrin β1 and the matrix
metalloproteinase, MT1-MMP (6).
Thus, GS is a potential target for the regulation of astrocyte
activation.
To date, however, only a limited number of studies
have investigated the changes in GS levels following SCI (7–9). It
is unknown whether these alterations in the GS levels contribute to
neuronal loss and are associated with glial activation following
SCI. In the present study, these issues were addressed by examining
the temporospatial cellular expression of GS following SCI using a
clinically relevant rat SCI model.
Materials and methods
Rat contusion injury and tissue
preparation
Female Sprague-Dawley rats (200–220 g) were used for
the induction of SCI. All animal experiments were approved by the
Institutional Animal Care and Use Committee of Nanjing Medical
University for the use of laboratory animals and performed in
accordance with the Guide for the Care and Use of Laboratory
Animals (National Research Council, 1996, USA). The rats were
randomly assigned to control and experimental groups. Contusive SCI
was performed as described previously (6,10,11).
Briefly, the rats were anesthetized by pentobarbital sodium (50
mg/kg; intraperitoneal injection) and the spinal cords were exposed
by laminectomy at the thoracic 9 (T9) level. Body temperature was
maintained at 37°C during anesthesia. Following the exposure of the
dorsal surface of the spinal cord, with the dura remaining intact,
a weight-drop injury was performed by dropping a 10-g rod
(diameter, 2.5 mm) from a height of 25 mm using the New York
University impactor (purchased from New York University
Neurosurgery Laboratory, New York, NY, USA). Sham-operated rats
were laminectomized but received no contusion. The rats were
perfused transcardially with 100 ml 0.9% saline followed by 200 ml
4% paraformaldehyde at day 1 or week 1 post-injury. Following
perfusion, the entire spinal cord was removed. The spinal cord
tissue (cervical to sacral) was separated into 5-mm segments.
Frozen transverse sections (20-μm thick) were obtained using a
cryostat (Leica CM1900; Leica Microsystems, Inc., Nussloch,
Germany) and thaw-mounted on poly-L-lysine-coated slides (Sigma,
St. Louis, MO, USA). The results are representative of sections
from 3 male and 3 female rats.
Immunofluorescence double-labeling
The immunofluorescence double-labeling method was
performed as described previously (12). Briefly, the sections were
premobilized and blocked with 0.3% Triton X-100 or 3% normal goat
serum in 0.01 M PBS for 30 min at room temperature (RT). A mixture
of GS rabbit polyclonal (1:2,000; Abcam, Cambridge, UK) and mouse
monoclonal antibodies were applied to the sections overnight at
4°C. The mouse monoclonal antibodies used in this experiment
included anti-β-III-tubulin (1:800; Sigma) to identify neurons,
anti-glial fibrillary acidic protein (GFAP; 1:800; Sigma) to
identify astrocytes, anti-RIP (1:25, a gift from Dr Scott R.
Whittemore, University of Louisville, KY, USA) to identify
oligodendrocytes, anti-OX42 (CD11b; 1:200; Chemicon, Temecula, CA,
USA) to recognize resting microglial cells and anti-CD68 (1:200;
R&D systems, Minneapolis, MN, USA) to recognize activated
microglia and macrophages. On the following day, the sections were
incubated with FITC-conjugated goat anti-rabbit and
TRITC-conjugated goat anti-mouse (1:100; Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA) antibodies. The sections
were washed, mounted and examined using the Olympus BX60 light
(Olympus, Center Valley, PA, USA) or Zeiss LSM 510 confocal
microscopes (Carl Zeiss AG, Oberkochen, Germany). Primary antiserum
omission and normal mouse and goat serum controls were used to
confirm the specificity of the immunofluorescent labeling.
Immunohistochemistry staining
Immunohistochemistry was performed using the
avidin-biotinylated peroxidase complex (ABC)
immunoperoxidase-diaminobenzidine tetrahydrochloride (DAB) method
according to the manufacturer’s instructions (Zymed; Invitrogen
Life Technologies, Carlsbad, CA, USA). Briefly, the sections were
incubated with mouse anti-GS antibodies (1:1,000; BD Biosciences,
Franklin Lakes, NJ, USA) and then reacted with biotinylated goat
anti-mouse IgG for 0.5 h at RT and subsequently with ABC for 0.5 h
at RT. The reaction product was revealed by incubating the slices
with DAB and H2O2 in substrate buffer for 15
min. Next, the sections were dehydrated, cleared and coverslipped.
The slides were examined using an Olympus BX60 light microscope.
The primary anti-serum omission and normal goat controls were used
to confirm the specificity of the immunohistochemical labeling.
Western blot analysis
Western blot analysis was performed as described
previously (6). Briefly, a 10-mm
cord segment containing the injury epicenter was removed and
homogenized by sonication in a lysis buffer. Protein (20 μg) from
the sample supernatant was measured using the BCA method and loaded
onto a 12% polyacrylamide gel, separated by SDS-PAGE and
transferred to PVDF membranes by electrophoresis. Following
membrane blocking, the mouse anti-GS (1:5,000; BD Biosciences) or
anti-GFAP antibodies (1:1,000; Sigma-Aldrich) were added to the
membrane and incubated at 4°C overnight. The membrane was washed
and incubated with secondary antibody goat anti-mouse IgG
conjugated with horseradish peroxidase (1:2,000; Amersham Pharmacia
Biotech, Arlington Heights, IL, USA) at RT for 2 h. Subsequent to
being washed, the membrane was analyzed by enhanced
chemiluminescence (Amersham Pharmacia Biotech).
GS activity measurement
GS activity in the tissue was measured using the
colorimetric assay as described previously (3) and then expressed as
c-glutamylhydroxamate (μM)/h/mg cell protein. The spinal cord
(10-mm section containing the injury epicenter) was obtained at
varying post-operative survival times. The tissue protein
concentrations were determined using the BCA protein estimation kit
(Pierce Biotechnology, Inc., Rockland, IL, USA).
Glutamate concentration analysis
The glutamate content in the spinal cord samples was
quantified by measuring the absorbance at 492 nm using the
Glutamate Colorimetric Assay kit (Genmed Scientific, Inc.,
Arlington, MA, USA) and expressed as glutamate (μM)/mg tissue
protein. A standard curve was constructed in each assay using known
concentrations of glutamate. The concentration of tissue glutamate
in the spinal cord (10-mm section containing the injury epicenter)
was estimated from the standard curve.
Statistical analysis
All data are expressed as mean ± SD. A one-way ANOVA
with Tukey’s HSD post hoc t-test was used to determine the level of
statistical significance. P<0.05 was considered to indicate a
statistically significant difference.
Results
Distribution of GS in the rat spinal
cord
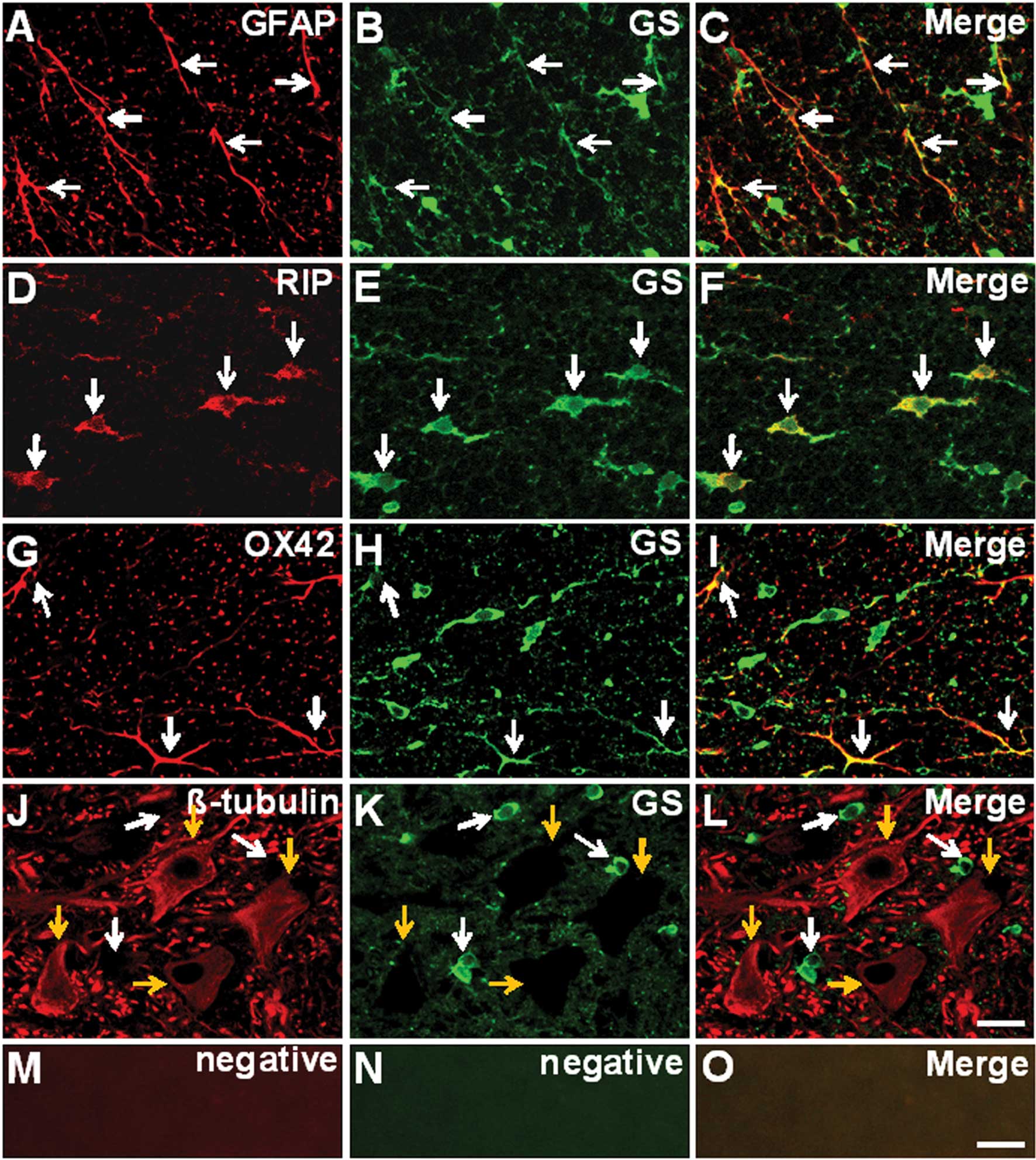
Immunofluorescence double staining was employed to
confirm the cellular localization of GS in the rat spinal cords.
Sections were obtained from an area 5 mm caudal to the epicenter of
the sham-operated rat spinal cord. GS-expressing glial cells,
including GFAP-positive astrocytes (Fig. 1A-C), RIP-positive oligodendrocytes
(Fig. 1D-F) and OX42-positive
microglia (Fig. 1G-I) were
identified. However, GS expression was not observed in
β-III-tubulin-positive neurons (Fig.
1J-L).
Glutamine synthetase activity and protein
levels following SCI
GS is the key glial-specificity enzyme that converts
glutamate to glutamine to affect the clearance of synaptic
glutamate (13). In the present
study, the correlation between GS activity and glutamate
concentration following SCI was analyzed. As shown in Fig. 2A, the concentration of glutamate
increased rapidly and reached a maximum of 1.26±0.08 fold at 1 h
compared with the sham-operated group (P=0.0048) post-injury. The
concentration returned to baseline rapidly at 4 h post-injury.
Fig. 2B demonstrates that a
significant decrease in GS activity occurred at 4 h post-injury
(P=0.018), then the GS activity increased, prior to returning to
the basal level at day 1 and then increasing to peak levels at day
7 post-injury (P=0.0037). The GS activity then remained at a high
level. SCI also caused a significant decrease in the GS expression
at 4 h (P=0.018) and day 1 (P=0.0079) post-injury. The GS protein
levels also revealed a similar time course change following SCI
(Fig. 2C and E). To test whether
the change in GS levels following SCI was associated with the
activity of glial cells, the correlation between the changes in GS
and GFAP, CD68 and PLP, the specific markers for astrocytes, active
microglia/macrophages and oligodendrocytes, respectively, was
determined (Fig. 2C-H). The
western blot analysis revealed that GFAP levels decreased 1 h
post-injury, decreased further at 4 h (p=0.018) and day 1
(P=0.0022) post-injury, then increased rapidly from day 3
(P=0.0099) and peaked at day 7 post-injury (P=0.000016). The GFAP
protein concentration remained at a high level from then onwards
(Fig. 2C and D). Fig. 2E and G revealed that CD68 was
almost undetectable in the sham-operated spinal cords and increased
significantly from 4 h following SCI (P=0.0011; Fig. 2F and G). However, the PLP
expression decreased gradually and was significantly decreased 1
day after SCI (P=0.0033; Fig. 2F and
H).
Distribution of GS in the injured spinal
cords
To determine the spatial distribution of GS
following SCI, immunochemistry staining for GS was performed at
days 1 and 7 post-injury, when the GS protein was minimally and
maximally expressed, respectively. In the sham-operated rats
(Fig. 3, left column), the GS
immunoreactivities revealed comparable basal levels in the glial
cells in the white and gray matter of the spinal cord. At day 1
post-injury, the GS immunoreactivities were markedly decreased in
the injured epicenter dorsal column and ventral horn gray and white
matter (Fig. 3B and E). However,
no significant difference was observed in the GS levels in the
region 5-mm caudal to the injury site (Fig. 3H and K). By contrast, the GS
immunoreactivities were markedly increased in the glial cells in
the white and gray matter 7 days after SCI (Fig. 3C and F). The strongest labeling was
observed at the lesion cavity and surrounding area. Specific GS
immunoreactivities were morphologically characteristic of activated
astrocytes. It appeared that certain cells with spherical
morphologies were derived from cells that infiltrated into the
lesion site. Furthermore, the increased GS immunoreactivities were
also observed throughout the entire length of the specimen (10 mm;
Fig. 3I and L).
Cellular localization of GS in the
injured spinal cord
The astrocyte response to injury was confined to the
immediate penumbra surrounding the lesion center. Astrocyte
reactivity was characterized by a cellular hypertrophy with a
process extension and increased production of intermediate
filaments, including GFAP (Fig.
4A). The upregulation of GS in the tissue surrounding the
lesion center was produced by GFAP-positive astrocytes (Fig. 4A-D). Since macrophages and
microglial cells express GS (14,15),
CD68 was used to identify the GS-positive infiltrative cells. As
revealed in Fig. 4E-H,
colocalization of GS and CD68 was observed in certain spherical
cells (indicated by arrows). Thus, these results indicate that the
infiltrative macrophages/activated microglia in areas close to the
injury site express GS.
Discussion
In the present study, changes in the GS levels in
the normal and injured rat spinal cords were systematically
analyzed. To the best of our knowledge, this is the first study to
demonstrate the temporospatial expression pattern of GS and its
cellular localization in the adult rat spinal cord following
SCI.
GS is expressed in glial cells, including
astrocytes, oligodendrocytes and microglia in the white and gray
matter in normal rat spinal cords. The enzyme is associated with
the regulation of glutamate concentration in the synaptic claft
(2) and therefore the expression
of GS in these supporting cells is vital to normal neural function
(13,16). Our recent study revealed that MMPs
are regulated by GS in astrocytes (6). Since MMPs in the CNS have been
implicated in synaptic plasticity, neuroprotection, oncogenesis and
oligodendrocyte differentiation (17), the effects of GS in the regulation
of neuronal function may be mediated by MMPs. GS may exert its
effect on astrocytes and Schwann cell differentiation by regulating
glutamate concentration (2,18).
These observations indicate that GS may play roles in the
differentiation of oligodendrocytes and astrocytes. However, the
function of GS in the microglia is not well documented.
GS is a key glutamate-metabolizing enzyme and the
removal of extracellular glutamate is dependent on the expression
level of this enzyme in glial cells (4,19).
In the present study, the activity and expression of GS were
initially maintained at low levels following SCI, which was
consistent with the marked elevation of glutamate at the injury
site. The downregulation of GS and the increased levels of
glutamate may be caused by the marked destruction of neuronal and
glial cells, including astrocytes and oligodendrocytes, in the
acute stage following SCI. On the other hand, the initial
inflammation triggered by SCI is mediated by the cytokines and
their receptors, including TNF-α and its receptor (20,21).
These cytokines induce neural excitotoxicity in glial cells
(22) by suppressing GS expression
in astrocytes (3) and may explain
why an increase in active microglia/macrophages at the early stages
subsequent to SCI did not induce an increase in GS, despite the
cells expression of GS. Thus, according to our previous study,
these results indicate that low levels of GS and a robust induction
of cytokine expression at early stages following SCI induces cell
death by excitotoxicity. However, the mechanism by which GS is
downregulated during the initial activation of the glial cells is
largely unknown.
Following CNS injury, astrocytes undergo a
transformation from a quiescent to a reactive state, in a process
known as dedifferentiation (23).
Previous studies have demonstrated that GS is associated with
gliosis following injury (8,24)
and that downregulation of GS is associated with the activation or
dedifferentiation of astrocytes (25). However, the role of GS in astrocyte
activation remains unclear. Our recent study demonstrated that the
downregulation of GS promotes the migration of astrocytes, a
process mediated by the upregulation of MT1-MMP and downregulation
of integrin β1 (6). The results
suggested that the temporal downregulation of GS in astrocytes at
the early stages of SCI is a promising strategy for SCI treatment,
which induces astrocytic activation and migration in order to
restrict the expansion of the inflammatory and lesional areas.
In the central nervous system, the importance of GS
in glutamate metabolism has been largely studied with respect to
excitotoxicity and/or glutamatergic neurotransmission between
neurons and astrocytes (26). The
present study revealed that GS is also expressed in
oligodendrocytes in the spinal cord. However, the function of GS in
these cells has not been clearly described. A previous study
demonstrated that GS is not only a metabolically relevant enzyme,
but that it also regulates Schwann cell differentiation and
promotes myelination by regulating the glutamate concentration
(18). These observations may aid
further studies in revealing the functional significance of GS in
oligodendrocytes and during the development and degeneration of the
central nervous system.
In the present study, macrophages and activated
microglia were found to invade the lesion sites 7 days post-injury
and exhibit GS-positive immunoreactivities. These results indicate
that, in addition to their recognized neurotoxic properties in
inflammation following SCI, these cells may exhibit specific
neuroprotective properties, which partly compensate for the
inhibition of astrocytic function. By contrast, microglia express
glutaminase, the enzyme which catalyzes the conversion of glutamate
from glutamine. A previous study reported that TNF-α induced
neurotoxicity via glutamate release from activated microglia
(27). Therefore, the function of
GS in these cells is complex. The clarification of the correlation
between GS and inflammation or inflammatory cells must be performed
as the function of GS in these cells is likely to form the basis
for any future clinical applications.
In conclusion, the present study demonstrated that
low levels of GS contribute to glutamate neurotoxicity at an early
stage following SCI and that the activation of glial cells leads to
changes in the GS levels. Understanding the function and mechanism
of GS is a novel strategy for studying the role of astrocytes and
microglia in the recovery of damaged tissue in SCI.
Acknowledgements
The current study was supported by grants from the
Natural Science Foundation of China (no. 81000527), the Natural
Science Foundation of Jiangsu Province (no. BK2010159) and the
Science and Technology Development Program of Huadong Sanitarium
(no. 201001). The authors would like to thank BiomedWorld
Bioscience Ltd., Co., for their editing of the manuscript.
References
|
1
|
Mills CD, Xu GY, McAdoo DJ and Hulsebosch
CE: Involvement of metabotropic glutamate receptors in excitatory
amino acid and GABA release following spinal cord injury in rat. J
Neurochem. 79:835–848. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Suárez I, Bodega G and Fernández B:
Glutamine synthetase in brain: effect of ammonia. Neurochem Int.
41:123–142. 2002.
|
|
3
|
Zou J, Wang YX, Dou FF, et al: Glutamine
synthetase down-regulation reduces astrocyte protection against
glutamate excitotoxicity to neurons. Neurochem Int. 56:577–584.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu B, Xu ZF, Deng Y, Liu W, Yang HB and
Wei YG: Protective effects of MK-801 on methylmercury-induced
neuronal injury in rat cerebral cortex: involvement of oxidative
stress and glutamate metabolism dysfunction. Toxicology.
300:112–120. 2012. View Article : Google Scholar
|
|
5
|
Silver J and Miller JH: Regeneration
beyond the glial scar. Nat Rev Neurosci. 5:146–156. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zou J, Wang YX, Mu HJ, et al:
Down-regulation of glutamine synthetase enhances migration of rat
astrocytes after in vitro injury. Neurochem Int. 58:404–413. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baldwin SA, Broderick R, Blades DA and
Scheff SW: Alterations in temporal/spatial distribution of GFAP-
and vimentin-positive astrocytes after spinal cord contusion with
the New York University spinal cord injury device. J Neurotrauma.
15:1015–1026. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Benton RL, Ross CD and Miller KE:
Glutamine synthetase activities in spinal white and gray matter 7
days following spinal cord injury in rats. Neurosci Lett. 291:1–4.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Romero-Alemán MM, Monzón-Mayor M, Yanes C
and Lang D: Radial glial cells, proliferating periventricular
cells, and microglia might contribute to successful structural
repair in the cerebral cortex of the lizard Gallotia
galloti. Exp Neurol. 188:74–85. 2004.
|
|
10
|
Lu HZ, Wang YX, Zou J, et al:
Differentiation of neural precursor cell-derived oligodendrocyte
progenitor cells following transplantation into normal and injured
spinal cords. Differentiation. 80:228–240. 2010. View Article : Google Scholar
|
|
11
|
Lü HZ, Xu L, Zou J, et al: Effects of
autoimmunity on recovery of function in adult rats following spinal
cord injury. Brain Behav Immun. 22:1217–1230. 2008.PubMed/NCBI
|
|
12
|
Hu JG, Fu SL, Zhang KH, et al:
Differential gene expression in neural stem cells and
oligodendrocyte precursor cells: a cDNA microarray analysis. J
Neurosci Res. 78:637–646. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eid T, Behar K, Dhaher R, Bumanglag AV and
Lee TS: Roles of glutamine synthetase inhibition in epilepsy.
Neurochem Res. 37:2339–2350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chrétien F, Vallat-Decouvelaere AV,
Bossuet C, et al: Expression of excitatory amino acid transporter-2
(EAAT-2) and glutamine synthetase (GS) in brain macrophages and
microglia of SIVmac251-infected macaques. Neuropathol Appl
Neurobiol. 28:410–417. 2002.PubMed/NCBI
|
|
15
|
Caldani M, Rolland B, Fages C and Tardy M:
Glutamine synthetase activity during mouse brain development.
Experientia. 38:1199–1202. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Coulter DA and Eid T: Astrocytic
regulation of glutamate homeostasis in epilepsy. Glia.
60:1215–1226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moore CS and Crocker SJ: An alternate
perspective on the roles of TIMPs and MMPs in pathology. Am J
Pathol. 180:12–16. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saitoh F and Araki T: Proteasomal
degradation of glutamine synthetase regulates schwann cell
differentiation. J Neurosci. 30:1204–1212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shaked I, Ben-Dror I and Vardimon L:
Glutamine synthetase enhances the clearance of extracellular
glutamate by the neural retina. J Neurochem. 83:574–580. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan P, Liu N, Kim GM, et al: Expression of
the type 1 and type 2 receptors for tumor necrosis factor after
traumatic spinal cord injury in adult rats. Exp Neurol.
183:286–297. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang XF, Huang LD, Yu PP, et al:
Upregulation of type I interleukin-1 receptor after traumatic
spinal cord injury in adult rats. Acta Neuropathol. 111:220–228.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie Z, Smith CJ and Van Eldik LJ:
Activated glia induce neuron death via MAP kinase signaling
pathways involving JNK and p38. Glia. 45:170–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lang B, Liu HL, Liu R, Feng GD, Jiao XY
and Ju G: Astrocytes in injured adult rat spinal cord may acquire
the potential of neural stem cells. Neuroscience. 128:775–783.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Politis MJ and Miller JE: Post-traumatic
alterations in glutamine synthetase activity in peripheral and
central nerves. Brain Res. 359:183–186. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Egnaczyk GF, Pomonis JD, Schmidt JA, et
al: Proteomic analysis of the reactive phenotype of astrocytes
following endothelin-1 exposure. Proteomics. 3:689–698. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levy HL: Metabolic disorders in the center
of genetic medicine. N Engl J Med. 353:1968–1970. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takeuchi H, Jin S, Wang J, et al: Tumor
necrosis factor-alpha induces neurotoxicity via glutamate release
from hemichannels of activated microglia in an autocrine manner. J
Biol Chem. 281:21362–21368. 2006. View Article : Google Scholar : PubMed/NCBI
|