Introduction
Breast cancer is the leading cause of cancer-related
mortality in the female population and chemotherapy alone or
combined with surgery currently forms the mainstay of clinical
treatment. In anthracycline-containing regimens, doxorubicin (ADR)
is considered as the main choice in chemotherapy for the treatment
of breast cancer since it effectively reduces the annual
probability of recurrence and mortality, particularly for high-risk
breast cancer. However, acquired resistance to ADR currently forms
a major obstacle to successful treatment.
The causes of cancer-specific drug resistance are
currently believed to be associated with epigenetic gene changes,
including DNA methylation (1–3).
Extensive studies have indicated the existence and importance of
another mechanism of non-mutational regulation of gene function,
which is mediated by microRNAs (miRNAs). miRNAs are non-coding RNAs
that mainly repress post-transcriptional gene expression, causing
the inhibition or degradation of mRNA translation (4). miRNA-200c, a member of the miRNA-200
family, has been shown to be downregulated in a variety of human
cancer types (5–7). A study by Cochrane et al
reported that chemosensitivity to paclitaxel was significantly
increased following the transfection of miRNA-200c in endometrial
cancer cells (8). Similarly, Ceppi
et al observed that the upregulation of miRNA-200c restored
the sensitivity of non-small cell lung cancer (NSCLC) cells to
cisplatin and cetuximab (9). These
results indicate that miRNA-200c is important in inhibiting the
development of drug resistance in cancer cells. miRNA-200c is also
aberrantly expressed in breast cancer cells with an ADR-resistant
phenotype (10), indicating that a
decrease in miRNA-200c expression is linked to ADR resistance in
breast cancer cells. However, the significance and precise
molecular mechanism by which this occurs remains unclear.
Phosphatase and tensin homolog (PTEN) is a
dual-specificity phosphatase, dephosphorylating lipid and protein
substrates, and is important in the suppression of tumor cell
proliferation and cell migration, signaling and apoptosis (11). Downregulation or mutation of the
PTEN gene is frequently observed in numerous types of human cancer,
including breast, prostate, brain, melanoma and glioma. Recently,
in a study aimed to investigate the acquired resistance mechanisms
to cetuximab in NSCLC, Kim et al demonstrated that PTEN
protein levels were decreased in cetuximab-resistant cell lines
(HCC827-CR), leading to the activation of phosphoinositide 3-kinase
(PI3K)/Akt signaling, while the upregulation of PTEN significantly
reduced Akt activity and restored drug sensitivity, indicating that
PTEN downregulation promoted the acquired resistance of NSCLC to
cetuximab by activation of PI3K/Akt signaling (12). A previous study has shown that PTEN
levels are regulated by E-cadherin, which is one of the most
important cell adhesion molecules in epithelial cells and
considered to function downstream of miRNA-200c. Restoration of
miRNA-200c activity directly targets and downregulates zinc finger
E-box-binding homeobox 1 (ZEB1), thereby increasing E-cadherin
expression and subsequently leading to EMT repression (13). In addition, the loss of E-cadherin
in ovarian cancer cells downregulated PTEN expression via
β-catenin-mediated Egr1 regulation, contributing to the activation
of PI3K/Akt signaling and cell proliferation (14). On the basis of these previous
findings, we hypothesize that downregulation of
miRNA-200c-dependent E-cadherin/PTEN signaling may lead to the
activation of PI3K/Akt, which subsequently regulates the ADR
resistance of breast cancer cells.
In this study, we aimed to demonstrate that
miRNA-200c inhibits the acquired resistance of breast cancer cells
against ADR via inactivation of the PI3K/Akt signaling pathway.
Upregulation of miRNA-200c results in an increase in the expression
of E-cadherin through inhibition of ZEB1 and a decrease in
phosphorylated Akt levels by upregulation of PTEN. By contrast,
miRNA-200c-upregulated PTEN levels are reduced when cells are
co-transfected with E-cadherin siRNA, leading to re-activation of
the PI3K/Akt signaling pathway. Furthermore, decreased Akt activity
increases the sensitivity of breast cancer cells to ADR. Our
results indicate that an interplay between the miRNA-200c assembly
and Akt signaling exists, which may provide important insights into
the role of miRNA-200c in ADR resistance.
Materials and methods
Cell culture
The human breast cancer MCF-7 and MCF-7/ADR
(resistant to ADR) cell lines were obtained from the Hebei Province
Cancer Institute (Hebei, China) and cultured in RPMI-1640 medium
supplemented with 10% fetal calf serum (Gibco BRL, Grand Island,
NY, USA) in a humidified atmosphere containing 5% CO2 at
37°C. For MCF-7/ADR cells, ADR (Sigma, St. Louis, MO, USA) was
additionally added at a final concentration of 1 mg/l to maintain
the resistance phenotype until one week prior to the
experiments.
Cell transfection
MCF-7/ADR cells were plated in 6-well plates
(4×105 cells/well) and transfected with 20 nM of either
miRNA-200c precursor (Applied Biosystems, Carlsbad, CA, USA) or a
mixture of miRNA-200c precursor and E-cadherin siRNA (20 nM,
reference sequence: 5′-CAGACAAAGACCAGGACUA-3′, Genepharma,
Shanghai, China) using Lipofectamin™ 2000 transfection reagent and
Opti-MEM I-reduced serum medium (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer’s instructions. Cells transfected
with scrambled RNA oligonucleotide served as a control. At 24 h
post-transfection, cells transfected with miRNA-200c precursor were
harvested for drug sensitivity assay and quantitative real-time
polymerase chain reaction (qRT-PCR).
Measurement of cell sensitivity to
ADR
The MTT
[3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide]
assay was used to determine drug sensitivity. MCF-7 and MCF-7/ADR
cells with cell transfection or treatment with Akt inhibitor
LY294002 were plated in 96-well plates at a concentration of
7×103 viable cells/well and cultured overnight. On the
following day, medium was replaced with fresh medium containing
different concentrations of ADR (final concentration: 0.1, 1, 10
and 100 μg/ml) and cells were incubated for 48 h. Cells were then
cultured for 4 h with 20 μl of MTT (5 mg/l, Sigma) and 150 μl of
DMSO. Absorbance was read at 490 nm using a microplate
spectrophotometer (Bio-Rad Laboratories, Hercules, CA, USA). The
IC50 value, defined as the drug concentration at which cell
survival was reduced to 50%, was assessed by the relative viability
curve according to the absorbance of MTT.
qRT-PCR analysis for miRNA-200c
expression
Total RNA was extracted from cells using TRIzol
(Invitrogen) according to the manufacturer’s instructions. After 1
μg of total RNA was reverse transcribed into cDNA, qRT-PCR was
performed using SYBR-Green PCR mastermix (Invitrogen). The primers
were synthesized by Shenggong Biotech Company Ltd. (Shanghai,
China) and the sequences of the miRNA-200c primers were: forward,
5′-GGTAATACTGCCGGGTAAT-3′ and reverse, 5′-CAGTGCTGTCGTGAGT-3′. The
sequences of the U6 primers were: forward,
5′-GCTTCGGCAGCACATATACTAAAAT-3′ and reverse,
5′-CGCTTCACGAATTTGCGTGTCAT-3′. PCR was performed using a thermal
cycler (Rotor-Gene 3000, Corbett Research, Australia) and the
conditions were as follows: denaturation at 95°C for 5 min,
followed by 40 cycles for 10 sec at 95°C, 20 sec at 60°C and 20 sec
at 78°C. The level of miRNA-200c was calculated using the
2−ΔCt method, where ΔCt was the difference in threshold
cycles for target and reference =
CtmiRNA-200c-CtU6 and expressed by the fold
changes relative to MCF-7/ADR.
Western blot analysis
MCF-7 and MCF-7/ADR cells with cell transfection or
treatment with Akt inhibitor LY294002 were collected and
homogenized in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl,
0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.02% sodium
azide, 100 mg/l PMSF, 1 mg/l aprotinin) and centrifuged at 14,000 ×
g for 10 min. The supernatant was collected and the total protein
content was determined using the Bradford assay. The proteins (50
μg/lane) were separated on 10% SDS-PAGE gels and transferred to
PVDF membranes (Bio-Rad). Membranes were blocked with 5% non-fat
milk and incubated overnight at 4°C with primary antibodies against
ZEB1, E-cadherin, PTEN, phospho-Akt (p-Akt, Ser473), total Akt and
β-actin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The
concentration of antibodies was 1:200 for ZEB1, E-cadherin, PTEN,
total Akt and p-Akt, and 1:1000 for β-actin. Following washing, the
membranes were incubated with infrared dye-labeled secondary
antibodies, IRDyeTM800DX-conjugated affinity-purified anti-mouse
IgG or IRDyeTM700DX-conjugated affinity-purified anti-rabbit IgG
(1:10000 dilution, LI-COR Biosciences, Lincoln, NE, USA). Target
bands were visualized using the Odyssey® Infrared
imaging system (version 3.0, LI-COR Biosciences). The results of
ZEB1, E-cadherin and PTEN were expressed as the ratio of the
density of specific bands compared with β-actin, while p-Akt was
normalized by total Akt.
Statistical analysis
Experiments were performed in triplicate. Data were
presented as the mean ± standard deviation (SD) and were analyzed
with the Student-Newman-Keuls t-test using SPSS 13.0 software
(Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant result.
Results
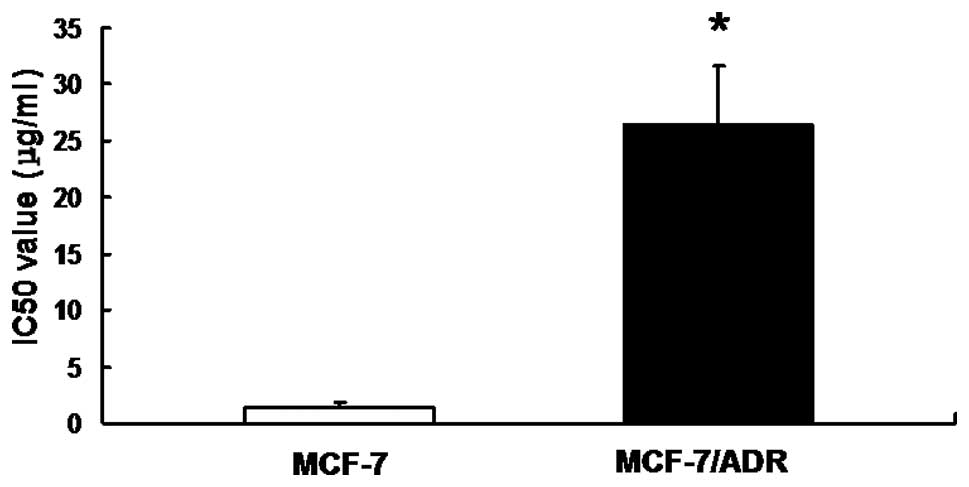
MCF-7/ADR cells demonstrated resistance
to ADR
To detect the drug resistance of MCF-7/ADR cells,
the in vitro sensitivity of MCF-7/ADR cells to ADR was
evaluated by MTT assay. Our results demonstrated that the IC50
value of MCF-7/ADR was 26.548±5.078 μg/ml, while the value for
MCF-7 alone was 1.364±0.494 μg/ml, suggesting that MCF-7/ADR cell
lines possessed a strong resistance to ADR (Fig. 1).
Restoration of miRNA-200c increased the
drug sensitivity of MCF-7/ADR cells to ADR
To determine the underlying mechanism for the
development of acquired ADR resistance in breast cancer cells, the
expression of miRNA-200c was initially detected using qRT-PCR. As
shown in Fig. 2A, MCF-7/ADR cells
exhibited decreased levels of miRNA-200c expression compared with
its parental MCF-7 cells. miRNA-200c precursor was transfected into
MCF-7/ADR cells and miRNA-200c expression was detected as well as
sensitivity to ADR. The results showed that transfection
signifcantly upregulated miRNA-200c expression in MCF-7/ADR cells
(Fig. 2B). Notably, the drug
sensitivity assay revealed that the IC50 value following
transfection of miRNA-200c precursor was markedly lower compared
with transfection with scrambled RNA oligonucleotide, indicating
that the sensitivity of MCF-7/ADR to ADR was restored (Fig. 2C). These data show that the drug
resistance of MCF-7/ADR cells was partially due to the decreased
expression of miRNA-200c.
Low miRNA-200c expression correlated with
low levels of E-cadherin protein and upregulation of ZEB1
expression
It is well established that cellular resistance to
chemotherapeutic drugs correlates with the loss of E-cadherin
expression (15) and recent
studies have demonstrated that miRNA-200c may be capable of
upregulating E-cadherin expression through direct targeting of its
ZEB1 transcriptional repressors, as a result of the
miRNA-200c-binding site being located within the ZEB1 3′
untranslated region (UTR) in breast cancer cells (13,16,17).
In the present study, we determined whether a negative correlation
between miRNA-200c and ZEB1 in MCF-7/ADR cells exists. Consistent
with previous studies (13), our
results demonstrated high levels of ZEB1 expression in MCF-7/ADR
cells with low levels of miRNA-200c expression. Since ZEB1 is a
potent repressor of E-cadherin, our results also showed that
E-cadherin protein was absent in MCF-7/ADR cells which did not
express miRNA-200c but expressed ZEB1 (Fig. 3A). To determine if translational
inhibition of ZEB1 restored the expression of E-cadherin, MCF-7/ADR
cells were transfected with miRNA-200c precursor. Fig. 3B demonstrates that the ectopic
upregulation of miRNA-200c expression levels efficiently reduced
the levels of ZEB1 protein in MCF-7/ADR cells. In addition, the
downregulation of ZEB1 in miRNA-200c-transfected MCF-7/ADR cells
was also accompanied by an increase in the level of E-cadherin
protein (Fig. 3B). Taken together,
our results indicate that the indirect upregulation of E-cadherin
may be involved in miRNA-200c-mediated drug sensitivity in
MCF-7/ADR cells.
Restoration of miRNA-200c suppressed Akt
signaling via E-cadherin-mediated PTEN regulation in MCF-7/ADR
cells
PTEN acts downstream of E-cadherin and directly
inhibits Akt signaling by inhibiting its phosphorylation. This
inhibition is important in tumor cell apoptosis and drug
resistance. Thus, we compared the levels of PTEN expression and Akt
phosphorylation between the two cell lines. As shown in Fig. 4A, MCF-7/ADR cells with depleted
E-cadherin levels exhibited decreased levels of PTEN expression and
an enhanced level of phosphorylated Akt. By contrast, forced
reintroduction of miRNA-200c precursor upregulated E-cadherin and
restored the expression of PTEN, eliminating Akt phosphorylation
(Fig. 4B). These results indicate
that E-cadherin levels positively correlate with PTEN and inversely
correlate with Akt phosphorylation. E-cadherin may additionally
inhibit Akt phosphorylation by positively regulating PTEN
expression. To further confirm the role of E-cadherin/PTEN/Akt
signaling in miRNA-200c-mediated drug resistance, we co-transfected
the miRNA-200c precursor and E-cadherin siRNA into MCF-7/ADR cells.
As shown in Fig. 4C, E-cadherin
siRNA inhibited the effects of the miRNA-200c precursor on the
changes of E-cadherin, PTEN and Akt phosphorylation. Taken
together, these data strongly suggest that miRNA-200c increases
PTEN expression by indirectly inducing E-cadherin expression, thus
leading to inactivation of Akt signaling by inhibiting its
phosphorylation.
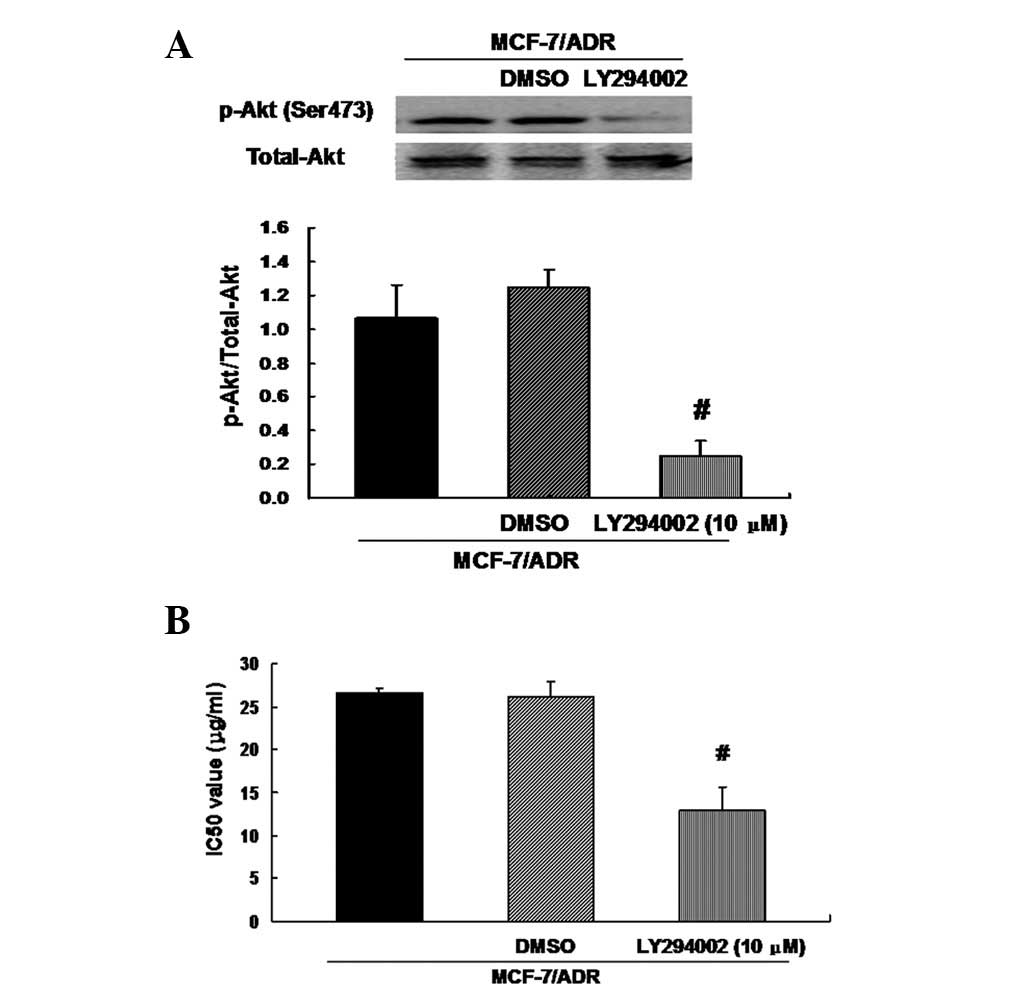
Inhibition of Akt signaling decreased the
resistance of MCF-7/ADR cells to ADR
The observation that upregulation of miRNA-200c
suppresses Akt phosphorylation and partially restores sensitivity
to ADR in MCF-7/ADR cells suggested the possibility that miRNA-200
inhibits drug resistance through inactivation of Akt signaling. To
test this hypothesis, we treated MCF-7/ADR cells with fresh medium
containing Akt inhibitor LY294002 (10 μM) for 24 h, and a control
with medium containing 0.1% dimethyl sulfoxide (DMSO) was used.
Treatment with LY294002 eliminated Akt phosphorylation in MCF-7/ADR
cells compared with the control (Fig.
5A). LY294002 additionally partially restored sensitivity of
MCF-7/ADR cells to ADR, as shown by the decreased IC50 values
(Fig. 5B). These data suggest that
miRNA-200c-mediated drug sensitivity of MCF-7/ADR cells is closely
correlated with the inactivation of Akt signaling.
Discussion
ADR exhibits a promising activity against numerous
types of cancer, including breast cancer, however, ADR resistance
of cancer cells remains a major obstacle for clinical treatment,
with its precise mechanism unclear. miRNA-200c has been shown to
inhibit tumorigenesis, including EMT, proliferation and metastasis.
Emerging evidence suggests that the loss of miRNA-200c expression
is a common event in the acquisition of cancer cell resistance to
chemotherapeutic drugs, indicating that miRNA-200c is also
important in the development of drug resistance of cancer cells. To
examine the effects of miRNA-200c on ADR resistance, we tested the
expression of miRNA-200c in breast cancer cells with ADR resistance
(MCF-7/ADR cells) and observed that miRNA-200c expression was
deceased in MCF-7/ADR cells compared with parental MCF-7 cells.
However, reintroduction of miRNA-200c by transfection with
miRNA-200c precursor into MCF-7/ADR cells markedly enhanced the
sensitivity of MCF-7/ADR cells to ADR, suggesting that an inverse
correlation between miRNA-200c expression and ADR resistance in
breast cancer cells may exist. Similarly, Kovalchuk et al
reported that miRNA-200c expression was lower in MCF-7 breast
cancer cells which possessed ADR resistance (10) and restoring miRNA-200c levels
increased ADR sensitivity through regulation of the p53 apoptotic
pathway in breast cancer MDA-MB-231 cells (18). These data demonstrate that the loss
of miRNA-200c is a critical determinant for the establishment of an
ADR-resistant phenotype in breast cancer.
In breast cancer cells with a mesenchymal phenotype,
overexpression of miRNA-200c enhances E-cadherin expression by
directly targeting and downregulating ZEB1 expression and
indirectly increasing acetylation of histone H3 at the E-cadherin
promoter, resulting in the repression of EMT (18). ZEB1 has been identified as a direct
transcriptional repressor of E-cadherin due to its abilitly to
directly bind E-box-like sequences in the E-cadherin promoter and
thereby repress E-cadherin (17).
Furthermore, E-cadherin is one of the key downstream regulators of
miRNA-200c contributing to EMT (16,19)
and is also important in inhibiting tumor invasion and
proliferation, as well as inducing cell apoptosis (20). It has been shown that the
expression of E-cadherin is lower in methotrexate-resistant human
HT29 colon cancer cells (15), and
restoration of E-cadherin expression levels increases the
sensitivity of cancer cells to anticancer agents, including
etoposide (21), taxol (22) and epidermal growth factor receptor
inhibitor (23,24). These findings indicate that
E-cadherin expression may be involved in the drug sensitivity of
tumor cells. In this study, we have demonstrated that the
expression of E-cadherin was decreased in MCF-7/ADR cells compared
with MCF-7 cells, while the level of ZEB1 expression was increased.
More importantly, upregulation of miRNA-200c expression
significantly repressed its direct target ZEB1 and restored
E-cadherin levels as well as the sensitivity to ADR. Supporting our
data, Park et al reported that miRNA-200c was identified as
an epithelial marker in NCI60 human cancer cell lines since it was
selectively expressed in only E-cadherin-positive and
vimentin-negative cancer cell lines that lack ZEB1 expression,
demonstrating a strong correlation between miRNA-200c and ZEB1 and
E-cadherin expression levels (13). Our data demonstrate that enforced
miRNA-200c expression in MCF-7 breast cancer cells causes the
repression of ZEB1, which subsequently increases the expression of
E-cadherin, leading to the increased sensitivity of breast cancer
cells to ADR.
E-cadherin has been shown to function as an
outside-in signaling receptor, transducing signals from the outside
to the inside of the cell. Therefore, examining how E-cadherin
affects the susceptibility of tumor cells to ADR within cells may
provide a better understanding with regard to the development of
miRNA-200c-mediated drug resistance in breast cancer cells. PTEN is
a dual-specificity phosphatase located in the cytoplasm which is
responsible for dephosphorylating lipid and protein substrates
(11). Lau et al reported
that loss of E-cadherin inhibited ovarian cancer cell growth by
repressing Egr1-mediated PTEN transcription (14). Li et al have shown that
restoring E-cadherin-mediated cell-cell adhesion improved PTEN
protein levels by increasing PTEN protein stability and inhibiting
degradation in human breast cancer cells (11). In agreement, we reported in the
present study that miRNA-200c-mediated depletion of E-cadherin in
MCF-7/ADR cells was associated with a decrease in PTEN levels as
evidence demonstrates that the upregulation of E-cadherin by
reintroduction of miRNA-200c precursor restored the expression of
PTEN. PTEN may inactivate the Akt signaling pathway by suppressing
Akt phosphorylation via dephosphorylating
phosphotidylinositol-(3,4,5)-triphosphate (the product of PI3K)
(25–28). Consistent with this finding, our
data showed that there was an increased expression of p-Akt level
in MCF-7/ADR cells, associated with low levels of PTEN. By
contrast, restoring miRNA-200c-mediated PTEN levels in MCF-7/ADR
cells by transfection with miRNA-200c precursor reduced p-Akt
expression. Specifically, knockdown of miRNA-200c-mediated
exogenous expression of E-cadherin reduced the expression levels of
E-cadherin and PTEN, and activated Akt phosphorylation again.
Therefore, our data suggest that dysregulation of PTEN expression
and Akt phosphorylation are crucial in E-cadherin-induced
intracellular signaling pathways.
In a study examining ovarian cancer, Lee et
al showed that the specific PI3K inhibitor LY294002 sensitized
cisplatin-resistant cell lines (OVCAR-3/CDDP) to cisplatin-induced
apoptosis and decreased cell viability, suggesting that activation
of the PI3K/Akt signaling may contribute to acquired cisplatin
resistance (29). In the present
study, we also observed the effects of Akt phosphorylation on ADR
resistance and showed that abolishing Akt phosphorylation by
LY294002 partially restored the sensitivity of MCF-7/ADR cells to
ADR. These data indicate that an inactivated Akt signaling pathway
may inhibit the development of drug resistance of breast cancer
cells.
In conclusion, we demonstrated that miRNA-200c is
important in the development of ADR resistance in breast cancer
cells. miRNA-200c increases the drug sensitivity of breast cancer
cells to ADR by inactivating the Akt pathway through the
E-cadherin-mediated upregulation of PTEN. This study provides novel
insights into the mechanisms of drug resistance of tumor cells and
highlights miRNA-200c as a new target to improve
chemosensitvity.
References
|
1
|
Roberti A, La Sala D and Cinti C: Multiple
genetic and epigenetic interacting mechanisms contribute to
clonally selection of drug-resistant tumors: current views and new
therapeutic prospective. J Cell Physiol. 207:571–581. 2006.
View Article : Google Scholar
|
|
2
|
Glasspool RM, Teodoridis JM and Brown R:
Epigenetics as a mechanism driving polygenic clinical drug
resistance. Br J Cancer. 94:1087–1092. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iwasa Y, Nowak MA and Michor F: Evolution
of resistance during clonal expansion. Genetics. 172:2557–2566.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Slaby O, Jancovicova J, Lakomy R, et al:
Expression of miRNA-106b in conventional renal cell carcinoma is a
potential marker for prediction of early metastasis after
nephrectomy. J Exp Clin Cancer Res. 29:902010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Philippidou D, Schmitt M, Moser D, et al:
Signatures of microRNAs and selected microRNA target genes in human
melanoma. Cancer Res. 70:4163–4173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karakatsanis A, Papaconstantinou I,
Gazouli M, Lyberopoulou A, Polymeneas G and Voros D: Expression of
microRNAs, miR-21, miR-31, miR-122, miR-145, miR-146a, miR-200c,
miR-221, miR-222, and miR-223 in patients with hepatocellular
carcinoma or intrahepatic cholangiocarcinoma and its prognostic
significance. Mol Carcinog. Dec 27–2011.(Epub ahead of print).
View Article : Google Scholar
|
|
8
|
Cochrane DR, Spoelstra NS, Howe EN,
Nordeen SK and Richer JK: MicroRNA-200c mitigates invasiveness and
restores sensitivity to microtubule-targeting chemotherapeutic
agents. Mol Cancer Ther. 8:1055–1066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ceppi P, Mudduluru G, Kumarswamy R, et al:
Loss of miR-200c expression induces an aggressive, invasive, and
chemoresistant phenotype in non-small cell lung cancer. Mol Cancer
Res. 8:1207–1216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kovalchuk O, Filkowski J, Meservy J, et
al: Involvement of microRNA-451 in resistance of the MCF-7 breast
cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther.
7:2152–2159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Z, Wang L, Zhang W, et al: Restoring
E-cadherin-mediated cell-cell adhesion increases PTEN protein level
and stability in human breast carcinoma cells. Biochem Biophys Res
Commun. 363:165–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim SM, Kim JS, Kim JH, et al: Acquired
resistance to cetuximab is mediated by increased PTEN instability
and leads cross-resistance to gefitinib in HCC827 NSCLC cells.
Cancer Lett. 296:150–159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lau MT, Klausen C and Leung PC: E-cadherin
inhibits tumor cell growth by suppressing PI3K/Akt signaling via
β-catenin-Egr1-mediated PTEN expression. Oncogene. 30:2753–2766.
2011.PubMed/NCBI
|
|
15
|
Selga E, Morales C, Noé V, Peinado MA and
Ciudad CJ: Role of caveolin 1, E-cadherin, Enolase 2 and PKCalpha
on resistance to methotrexate in human HT29 colon cancer cells. BMC
Med Genomics. 1:352008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hurteau GJ, Carlson JA, Spivack SD and
Brock GJ: Overexpression of the microRNA hsa-miR-200c leads to
reduced expression of transcription factor 8 and increased
expression of E-cadherin. Cancer Res. 67:7972–7976. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eger A, Aigner K, Sonderegger S, et al:
DeltaEF1 is a transcriptional repressor of E-cadherin and regulates
epithelial plasticity in breast cancer cells. Oncogene.
24:2375–2385. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tryndyak VP, Beland FA and Pogribny IP:
E-cadherin transcriptional down-regulation by epigenetic and
microRNA-200 family alterations is related to mesenchymal and
drug-resistant phenotypes in human breast cancer cells. Int J
Cancer. 126:2575–2583. 2010.
|
|
19
|
Hurteau GJ, Spivack SD and Brock GJ:
Potential mRNA degradation targets of hsa-miR-200c, identified
using informatics and qRT-PCR. Cell Cycle. 5:1951–1956. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Junxia W, Ping G, Yuan H, et al: Double
strand RNA-guided endogeneous E-cadherin up-regulation induces the
apoptosis and inhibits proliferation of breast carcinoma cells in
vitro and in vivo. Cancer Sci. 101:1790–1796. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sasaki CY, Lin HC and Passaniti A:
Expression of E-cadherin reduces bcl-2 expression and increases
sensitivity to etoposide-induced apoptosis. Int J Cancer.
86:660–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ferreira P, Oliveira MJ, Beraldi E, et al:
Loss of functional E-cadherin renders cells more resistant to the
apoptotic agent taxol in vitro. Exp Cell Res. 310:99–104. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Black PC, Brown GA, Inamoto T, et al:
Sensitivity to epidermal growth factor receptor inhibitor requires
E-cadherin expression in urothelial carcinoma cells. Clin Cancer
Res. 14:1478–1486. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Witta SE, Gemmill RM, Hirsch FR, et al:
Restoring E-cadherin expression increases sensitivity to epidermal
growth factor receptor inhibitors in lung cancer cell lines. Cancer
Res. 66:944–950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oki E, Baba H, Tokunaga E, et al: Akt
phosphorylation associates with LOH of PTEN and leads to
chemoresistance for gastric cancer. Int J Cancer. 117:376–380.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stassi G, Garofalo M, Zerilli M, et al:
PED mediates AKT-dependent chemoresistance in human breast cancer
cells. Cancer Res. 65:6668–6675. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Myers MP, Pass I, Batty IH, et al: The
lipid phosphatase activity of PTEN is critical for its tumor
supressor function. Proc Natl Acad Sci USA. 95:13513–13518. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee S, Choi EJ, Jin C and Kim DH:
Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA
amplification contributes to cisplatin resistance in an ovarian
cancer cell line. Gynecol Oncol. 97:26–34. 2005. View Article : Google Scholar : PubMed/NCBI
|