Introduction
Chromium (Cr) exists in the workplace mainly in two
valence states: hexavalent Cr [Cr(VI)] and trivalent Cr [Cr(III)]
(1). Cr(VI) has extensive
applications in diverse industries, including artistic paints,
electroplating and stainless steel welding; while Cr(III) is often
used as a micronutrient or a dietary supplement (2). Cr(III) is known to be less toxic than
Cr(VI), due to its inability to pass through transporters residing
within the cell membrane (3).
Cr(VI) and its compounds are widely known to cause contact
dermatitis and nasal perforation, as well as carcinogenic effects
in humans and animals (4). A large
number of workers are exposed to Cr due to its widespread use.
Occupational exposure to Cr(VI) mainly occurs via long-term chronic
inhalation, and it is estimated that >700,000 workers are
potentially exposed to high levels of Cr(VI) in China (5). Although attempts have been made to
explore the toxicity of heavy metals in recent years, Cr(VI) has
received little attention in comparison to Zn, Cu, Pb and Cd.
In addition to the production of ATP, mitochondria
are involved in numerous other cellular functions, including
metabolic regulation signaling and cell growth, differentiation and
death (6). The mechanisms by which
mitochondrial damage affects cell function are well documented.
Numerous studies have demonstrated that Cr(VI) is capable of
inducing a variety of types of DNA damage, including single-strand
breaks, alkali-labile sites and DNA-protein crosslinks (DPCs) in
various cells in cell culture and in vivo(7). The formation of DPCs in target
tissues appears to be the direct and primary genotoxic effect of
Cr(VI) exposure. Welders (8),
chrome platers (9) and leather
tanners (10) have demonstrated
increased levels of DPCs in peripheral blood lymphocytes (PBLs). A
previous study supported the hypothesis that there is a strong
correlation between lymphocytic DPCs and Cr levels in red blood
cells (11), thus the lymphocytic
DPCs may be viewed as a biomarker of internal Cr(VI)
accumulation.
There is also evidence to suggest that Cr(VI) and
its compounds are genotoxic and may induce gene mutations (12,13).
Cr-DPCs are stable and ternary DNA adducts, which constitute a
significant class of Cr-related genetic lesions, are able to block
the normal processes of replication and transcription (14,15).
Additionally, it has been demonstrated that oxidative stress, which
occurs when reactive oxygen species (ROS) reach abnormally high
levels, is able to affect the formation of ≤50% of the DPCs
(16). ROS, including superoxide
(O2•−), hydroxyl radicals (HO•)
and non-radical molecules, such as hydrogen peroxide
(H2O2), are known to initiate peroxidative
cell damage (17). However, the
relative contribution of ROS to Cr(VI)-induced DPCs is unclear and
is the subject of debate.
Vitamin C (vit C), also known as ascorbic acid, is a
water-soluble vitamin that has been demonstrated to be a key
antioxidant, which is capable of reducing metal-induced lipid
peroxidation and oxidative stress in animals (18). It is well known that vit C is an
important biological reducing agent in humans and animals, which is
capable of reducing Cr(VI) (19).
Therefore, supplementation of vitamins from external sources has
become necessary to prevent Cr(VI)-induced toxicity, including DNA
damage. A previous in vivo study demonstrated that vit C
significantly decreased the cytotoxicity and mutagenicity induced
by Cr(VI) in rats and guinea pigs (20). However, there is also evidence to
suggest that vit C may aggravate Cr(VI)-induced DNA damage by
increasing Cr-DNA binding and DNA strand breaks in
vitro(21). Therefore, this
raised the possibility that vit C may have different time-order
effects when antagonizing Cr(VI)-induced cytotoxicity and DNA
damage. Regardless of the evidence from cell culture and in
vivo experiments with regard to the protective effect of the
antioxidant, vit C, following exposure to Cr(VI), no studies have
been conducted to date to demonstrate the time-order effects of vit
C on Cr(VI)-induced mitochondrial damage and DPC formation.
Additionally, although there have been a large amount of studies
concerned with understanding the molecular mechanisms associated
with Cr(VI) exposure, the relative contributions of mitochondrial
dysfunction and DNA damage to Cr(VI) toxicity are an area that as
yet remains relatively unexplored. In the present study, we aimed
to investigate the time-order effects of vit C on Cr(VI)-induced
mitochondrial damage and DPCs in cultured rat PBLs. We also
conducted a mechanistic study to demonstrate the role of ROS and
p53 in Cr(VI)-induced mitochondrial damage and DPC formation.
Materials and methods
Animals
Male Sprague-Dawley (SD) 8-week-old rats were
obtained from the animal center of Central South University
(Changsha, Hunan, China). The rats were clinically normal, without
infection or inflammation, and were housed in clean cages with free
access to water and food. The animals were maintained in a 12-h
light/dark cycle at a temperature of 22±2°C and a humidity of
55±5%. The study was conducted with the approval of the Animal Care
and Use Committee of Central South University.
Materials
Vit C, 3-(4,5-dimethylthiazol-2yl-)-2,5-diphenyl
tetrazolium bromide (MTT), Hoechst 33258, Coomassie Brilliant Blue
(G-250) and rhodamine 123 (Rh123) were purchased from Sigma (St.
Louis, MO, USA). RPMI-1640 culture medium, fetal bovine serum (FBS)
and trypsin-EDTA (0.25%) were obtained from Gibco-BRL
(Gaithersburg, MD, USA). Potassium dichromate
(K2Cr2O7) was obtained from
Changsha Chemical Reagents Company (Changsha, Hunan, China) and was
used as the standard reagent. All other chemicals and solvents were
of analytical grade, HPLC grade or the optimum pharmaceutical
grade.
Preparation of rat PBLs
The preparation of rat PBLs was performed as
previously described with slight modifications (22,23).
A peripheral blood sample (20 ml) was drawn from SD rats and
collected in a heparinized tube. As the lymphocytes mainly
comprised of mononuclear cell fractions, we immediately (within 2
h) isolated the PBLs by density gradient centrifugation (1,200 × g,
20 min) using Gradiaol L (Aqua-Medic, Kobylnica, Poland). The
lymphocytes were then collected, and washed twice with 0.9% NaCl
solution and twice with transport solution (20 mM HEPES, 150 mM
NaCl, 5 mM KCl, 5 mM MgSO4, 1.2 mM
KH2PO4, 2.5 mM CaCl2, 2 mM
pyruvate, pH 7.4). An adequate volume of transport solution was
added to the isolated PBLs to obtain the density of 106
cells/ml.
Evaluation of cell viability
An MTT assay was performed to evaluate the cell
viability as previously described; however, with slight
modifications (24). The PBLs were
seeded in each well of a 96-well plate with 100 μl medium
containing 104 cells.
K2Cr2O7, vit C and the different
combinations of the two chemicals of indicated final concentrations
were added to each well, respectively. Control cells and medium
controls without cells received dimethylsulfoxide (DMSO) only.
Following incubation at 37°C for the indicated time period, the
PBLs were treated with 5 μl 5 mg/ml MTT for an additional 4 h at
37°C, and then lysed in phosphate-buffered saline (PBS, pH 7.4)
containing 20% sodium dodecyl sulfate (SDS) and 50%
N,N-dimethylformamide (pH 4.5). The absorbance was read at 570 nm
on a Versamax multiwell enzyme-linked immunosorbent assay (ELISA)
reader (Molecular Devices, LLC, Sunnyvale, CA, USA).
Measurement of DPC formation
The assay for measuring DPC formation was based on
the binding of SDS to proteins and its lack of binding to DNA,
according to the K-SDS assay developed by Zhitkovich and Costa
(25). Protein-bound DNA is easily
separated from free DNA, as protein-linked DNA precipitates with
SDS, while free DNA remains in the supernatant when the cation is
altered from Na to K. This method provided a direct measurement of
DPC formation by analyzing the quantity of DNA in the SDS pellet.
Following chemical exposure, the PBLs were washed and then
resuspended in 100 μl ice-cold PBS. The suspensions were lysed with
500 μl lysis buffer [1% SDS, 20 mM Tris-HCl, 1 mM
phenylmethylsulfonyl fluoride (PMSF), pH 7.5] at −70°C overnight.
The frozen tubes were thawed at 65°C in a water bath for 10 min,
and then the DNA was sheared by passing 10 times through a syringe
with a 21-gauge needle (foam must be avoided). Then, 100 μl
precipitation solution (2.5 M KCl, 10 mM Tris-HCl, pH 7.4) was
added and vortexed for 10 sec. The tubes were incubated at 65°C for
10 min in a water bath, cooled on ice and then centrifuged at 6,000
× g for 5 min. Subsequently, the pellet was resuspended by adding 1
ml scavenging buffer solution (0.1 M KCl, 0.1 mM EDTA, 20 mM
Tris-HCl, pH 7.4) and heating for 5 min at 65°C. The heating step
was then repeated and the pellet was washed three times. The pellet
was resuspended in 500 μl scavenging buffer solution, heated to
65°C and then digested with 500 μl 0.4 mg/ml protease K at 50°C for
3 h. Subsequently, the pellet was incubated with 1 ml freshly
prepared fluorescent dye (Hoechst 33258) to the final concentration
of 200 mg/ml, for 30 min in the dark. A fluorescence
spectrophotometer with an excitation wavelength of 360 nm and an
emission wavelength of 450 nm was used to assay the DPC
coefficient. The DPC coefficient was calculated as the ratio of the
percentage of DNA involved in DPCs to the percentage of total DNA
(DNA involved in DPCs and the unbound fraction of DNA).
Measurement of malondialdehyde (MDA)
content
The PBLs were lysed using the Mammalian Cell Lysis
kit, which was purchased from Sigma-Aldrich (St. Louis, MO, USA).
The total protein content of the PBLs was isolated and the protein
concentrations were determined using the Bicinchoninic Acid (BCA)
Protein Assay kit (Sigma-Aldrich). The measurement of MDA content
was performed using the MDA Content Detection Assay kit (Jiancheng
Institute of Biotechnology, Nanjing, Jiangsu, China) according to
the manufacturer’s instructions.
Measurement of the open permeability
transition pore (PTP) rate
The PBLs were collected following chemical exposure
and were then processed for mitochondrial isolation as previously
described (26). The cell pellets
were resuspended with solution A (250 mM sucrose, 20 mM HEPES, 10
mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM
dithiothreitol, 0.1 mM PMSF, pH 7.5). The PBLs were homogenized by
syringe homogenization and then centrifuged twice at 750 × g for 10
min at 4°C. The supernatants were collected and centrifuged at
10,000 × g for 15 min at 4°C, to obtain the mitochondrial pellets.
Mitochondrial protein concentrations were estimated using Coomassie
Brilliant Blue (G-250) according to the method developed by
Bradford (27). The opening of the
PTP was determined using a fluorescence spectrophotometer, with a
wavelength of 520 nm for the excitation and emission, at 25°C.
Measurement of the mitochondrial membrane
potential (MMP)
According to the method previously described by
Emaus et al(28), with
certain modifications, the MMP was monitored by the changes in the
fluorescence of a specific fluorescent cationic dye, Rh123, the
uptake of which by mitochondria is strongly dependent on the
transmembrane potential. The absorbance was recorded by a
fluorescence spectrophotometer at an excitation wavelength of 495
nm and an emission wavelength of 535 nm, at 25°C. For each test,
Rh123 (final concentration, 30 μM) was added to the mitochondrial
suspension.
Measurement of ROS production
The mitochondrial ROS production was determined
spectrofluorimetrically, by detecting the fluorescence intensity of
2′,7′-dichlorofluorescein (DCF), the oxidized product of the
membrane permeable fluoroprobe 5-(and
6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
(CM-H2DCFDA; Molecular Probes, Carlsbad, CA, USA).
Briefly, following chemical exposure, 2×106 PBLs were
collected and incubated with 10 μM CM-H2DCFDA solution
for 1 h at 37°C, in the dark. Fluorescence was measured with a
fluorescence spectrometer with an excitation wavelength of 488 nm
and an emission wavelength of 535 nm. The cellular ROS levels were
considered to be directly proportional to fluorescence
intensity.
Western blot analysis
Western blot analysis was performed using the
WesternBreeze Chemiluminescent Immunodetection kit (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer’s
instructions. Sample proteins (40 μg) were separated by
electrophoresis on 10% SDS polyacrylamide gels (SDS-PAGE) and then
transferred onto polyvinylidene difluoride (PVDF) membranes. The
membranes were incubated with primary antibodies overnight at 4°C,
blocked with 4% non-fat milk and then incubated for 1 h with
secondary antibodies at room temperature. The membranes were
developed with the detection system and exposed to films. The
primary antibody for p53 (PAb 240) (ab26) was purchased from Abcam
(Cambridge, MA, USA) and β-actin was purchased from Cell Signaling
Technology, Inc. (no. 4967; Danvers, MA, USA).
Statistical analysis
Statistical analysis was performed using one-way
analysis of variance (ANOVA), in the Statistical Package for the
Social Sciences (SPSS) v15.0 software (SPSS, Inc., Chicago, IL,
USA), to assess the significance of the differences between groups.
The results are expressed as mean ± standard deviation and were
calculated from quantitative data obtained from three replicate
experiments. P<0.05 was considered to indicate a statistically
significant result.
Results
Time-order effect of vit C on
Cr(VI)-induced inhibition of PBL viability
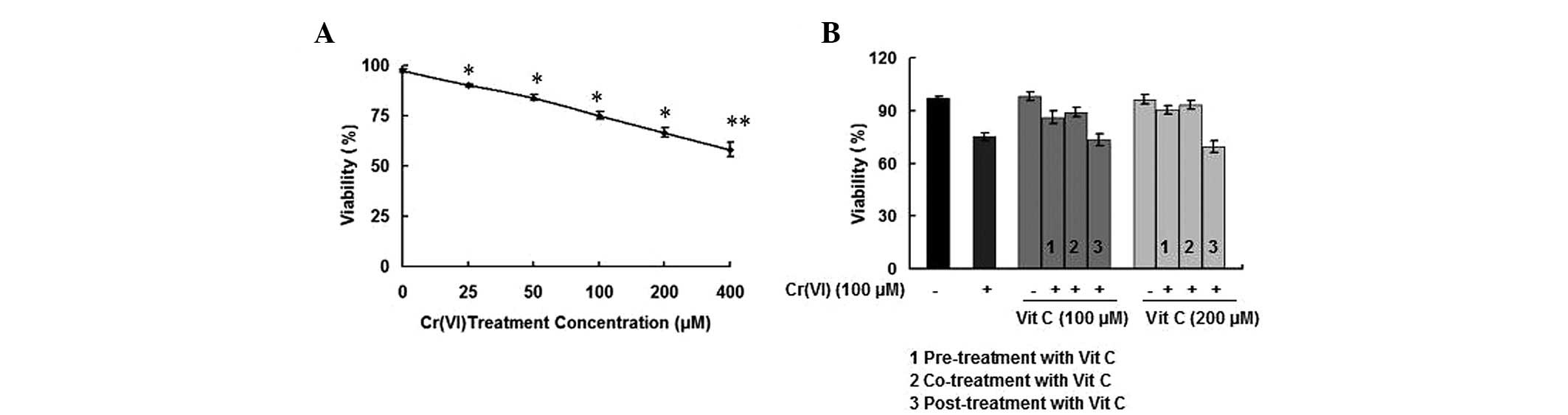
To investigate whether Cr(VI) affected the viability
of PBLs, the response of cells to different doses (25, 50, 100, 200
and 400 μM) of Cr(VI) was tested using the MTT assay. As shown in
Fig. 1A, Cr(VI) decreased the
viability of PBLs in a dose-response manner. A concentration of 100
μM Cr(VI) caused ~30% inhibition of cell viability, and this Cr(VI)
concentration was used for the following experiment. We then sought
to determine whether different time-order treatments of vit C with
Cr(VI) altered the inhibition of cell viability in the PBLs. In the
vit C alone treatment group, >95% of the cells were alive,
indicating that the concentrations of 100 and 200 μM vit C were not
the cytotoxic concentrations in PBLs following treatment for 6 h.
Both vit C pre- and co-treatment protected PBLs from the
deleterious effects of Cr(VI) toxicity, and resulted in higher
viability compared with treatment with Cr(VI) alone (P<0.05),
particularly in the 200-μM vit C groups. While co-treatment with
vit C and Cr(VI) demonstrated the optimal protective effect on cell
viability, vit C post-treatment did not rescue the decreased cell
viability compared with treatment with Cr(VI) alone (P>0.05)
(Fig. 1B). The results indicated
that when PBLs were exposed to Cr(VI) together with different time
orders of vit C treatments, vit C pre- and co-treatment exerted a
protective effect against Cr(VI)-induced inhibition of cell
viability.
Time-order effect of vit C on
Cr(VI)-induced DPC formation in PBLs
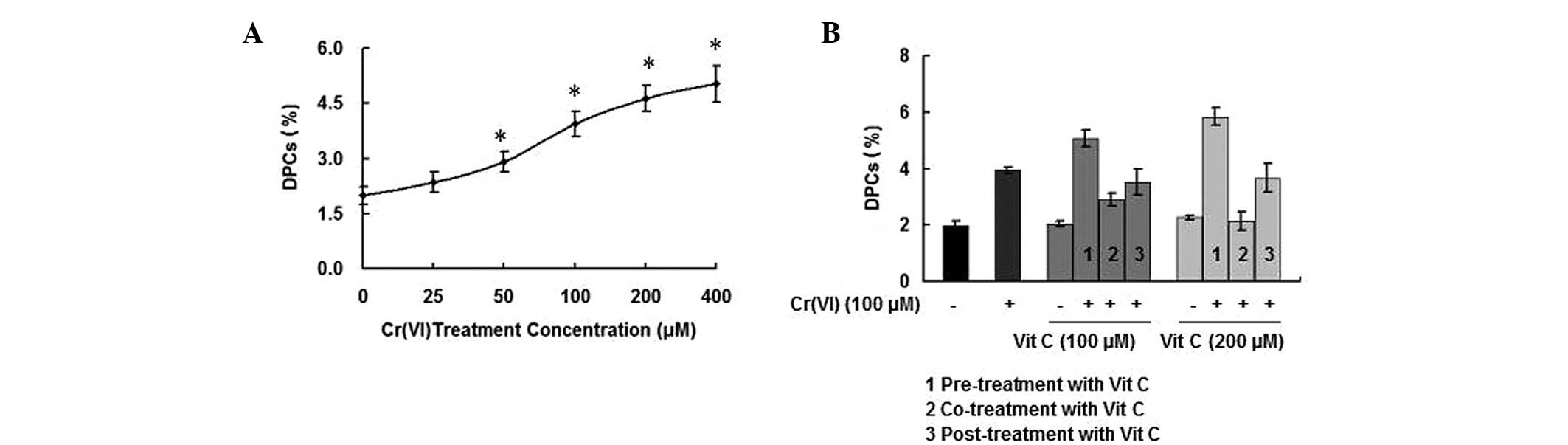
Previous studies on Cr(VI)-induced toxicity
suggested the potential utility of DPC lesions as biomarkers of
exposure to Cr(VI) and its compounds (29). Treatment of PBLs with various
concentrations of Cr(VI) (0, 25, 50, 100, 200 and 400 μM) for 6 h
resulted in a significant increase in DPCs, in a dose-dependent
manner (Fig. 2A). The DPC
coefficients of the treatment groups receiving vit C alone (100 and
200 μM) were not significantly different compared with that of the
control group (P>0.05). However, compared with the treatment
groups receiving vit C alone, more DPC formation occurred in the
Cr(VI) treatment groups that were pre-treated with vit C
(P<0.05). Additionally, while there was no significant
difference in DPC formation between the Cr(VI) treatment groups
that received vit C post-treatment and the treatment group that
received Cr(VI) alone (P>0.05), the combination treatment of
Cr(VI) with vit C demonstrated a protective effect against DPC
occurrence (P<0.05), particularly when the treatment
concentration of vit C was 200 μM (Fig. 2B). The results indicated that when
PBLs were exposed to Cr(VI) together with different time orders of
vit C treatments, only vit C co-treatment exerted a protective
effect against the Cr(VI)-induced increase in DPC formation.
Time-order effect of vit C on
Cr(VI)-induced mitochondrial damage
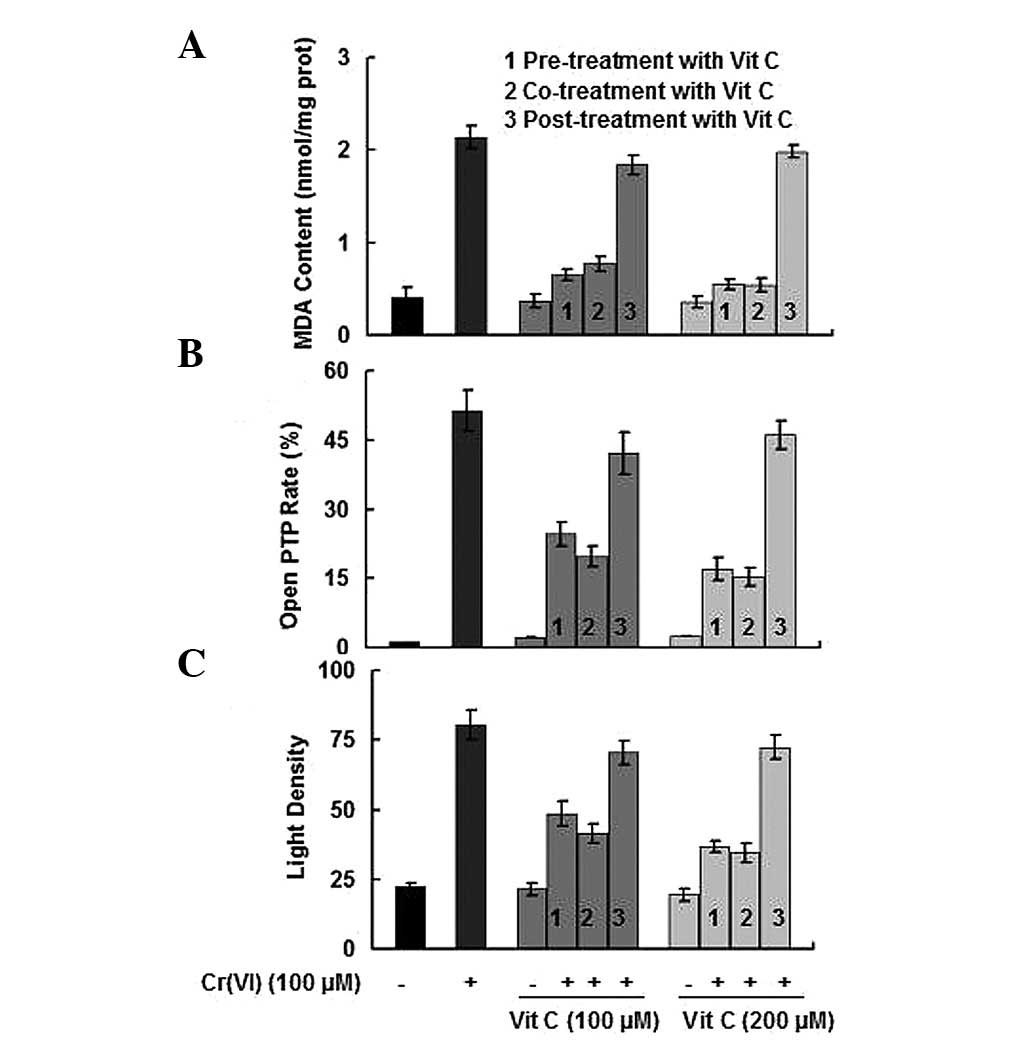
In addition to the opening status of the PTP and the
maintenance of the MMP, mitochondrial damage may also be evaluated
by the content of MDA (30). As
shown in Fig. 3, Cr(VI) treatment
resulted in a significant increase in the MDA content and the open
PTP rate; however, a decrease in MMP (signified by an increase in
the light density of Rh123; P<0.05). The results indicated that
when PBLs were exposed to Cr(VI) together with different time
orders of vit C treatments, while the Cr(VI) treatment groups that
received vit C post-treatment did not rescue mitochondrial damage,
vit C pre-treatment and co-treatment demonstrated a protective
effect against Cr(VI)-induced mitochondrial damage, particularly
when the treatment concentration of vit C was 200 μM.
Cellular ROS levels correlate with
Cr(VI)-induced mitochondrial damage
As ROS play a central role in Cr(VI)-induced
cytotoxicity in different types of cells, we measured the ROS
levels in PBLs exposed to various concentrations of Cr(VI) (25, 50,
100, 200 and 400 μM). By utilizing the oxidant-sensitive
fluorogenic probe, CM-H2DCFDA, we identified that Cr(VI)
stimulation induced significantly higher values of DCF fluorescence
in a dose-dependent manner compared with the control group
(P<0.05), suggesting a greater quantity of cellular ROS were
generated in the Cr(VI) treatment groups (Fig. 4A, upper panel). When detected under
the fluorescence microscope, we observed that Cr(VI) (100 μM)
induced a significantly higher level of fluorescence signals
compared with the control group (Fig.
4A, bottom panel). We then determined the ROS levels in the
groups of different time-order treatments of vit C with Cr(VI) in
PBLs. As shown in Fig. 4B, the
cells treated with 100 μM Cr(VI) and vit C exhibited up to a
~4-fold increase in ROS levels when compared with the control. The
ROS levels of the treatment groups receiving vit C alone (100 and
200 μM) were not significantly different (P>0.05). Compared with
the treatment groups receiving Cr(VI) alone, while the Cr(VI) plus
vit C post-treatment group demonstrated no significant change in
ROS levels (P>0.05), the Cr(VI) groups receiving vit C pre- and
co-treatment demonstrated significantly lower ROS levels
(P<0.05). As the changes in the ROS levels in the groups of
different time-order treatments of vit C with Cr(VI) are similar to
the changes in MDA content, open PTP rate, and MMP (Fig. 3), we concluded that cellular ROS
levels were correlated with Cr(VI)-induced mitochondrial
damage.
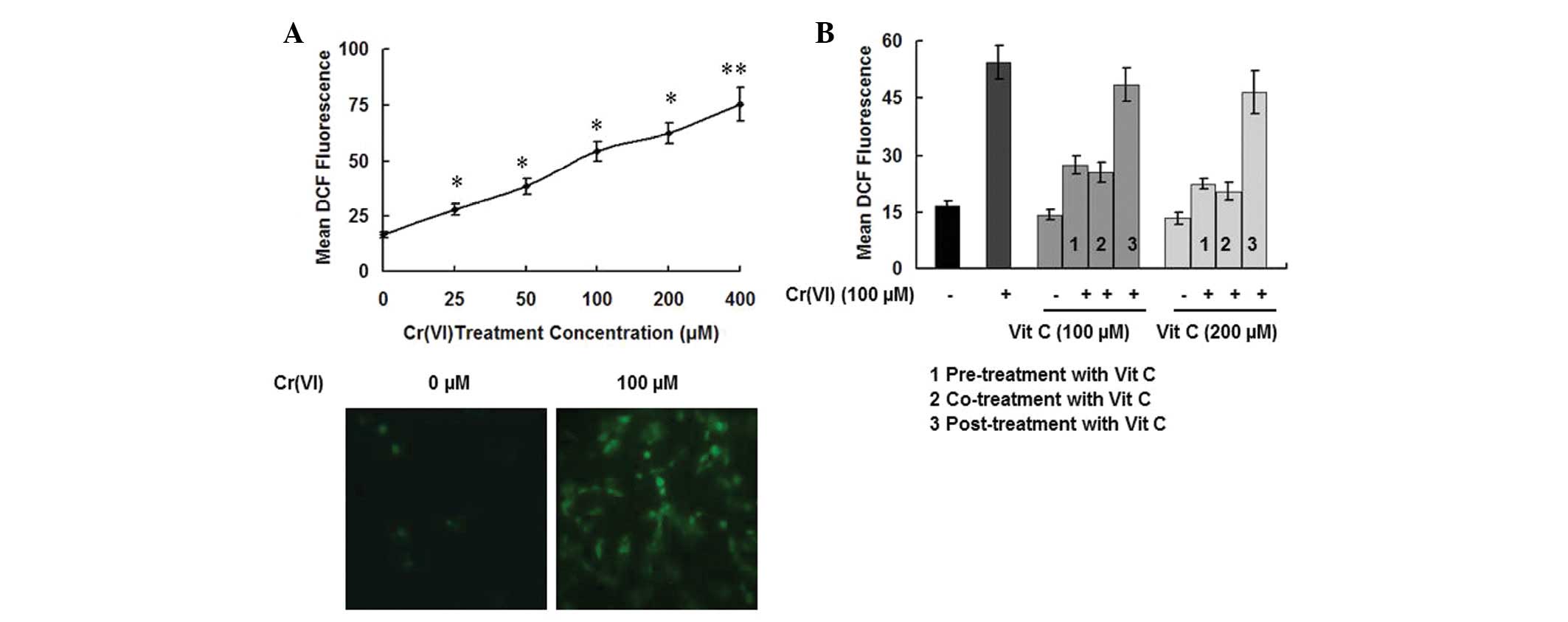 | Figure 4Time-order effect of vit C on
Cr(VI)-induced ROS accumulation in PBLs. (A) Cr(VI) induces ROS
accumulation. We measured the ROS levels in PBLs exposed to various
concentrations of Cr(VI) (25, 50, 100, 200 and 400 μM) by utilizing
the oxidant-sensitive fluorogenic probe CM-H2DCFDA. The
level of ROS production, which was considered to be directly
proportional to fluorescence intensity, was quantitated by flow
cytometry (upper panel) and was detected under a fluorescence
microscope (bottom panel). *P<0.05 and
**P<0.01 compared with the control group. (B)
Different time-order effects of vit C [pre-treatment with vit C for
2 h plus Cr(VI); co-treatment with Cr(VI) and vit C; and Cr(VI)
treatment for 2 h plus vit C post-treatment] on Cr(VI)-induced ROS
accumulation were detected. Two concentrations of vit C (100 and
200 μM) were employed. Data are expressed as the mean ± standard
deviation of three independent experiments. vit C, vitamin C;
Cr(VI), hexavalent chromium; PBL, peripheral blood lymphocyte; ROS,
reactive oxygen species; CM-H2DCFDA, 5-(and
6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate. |
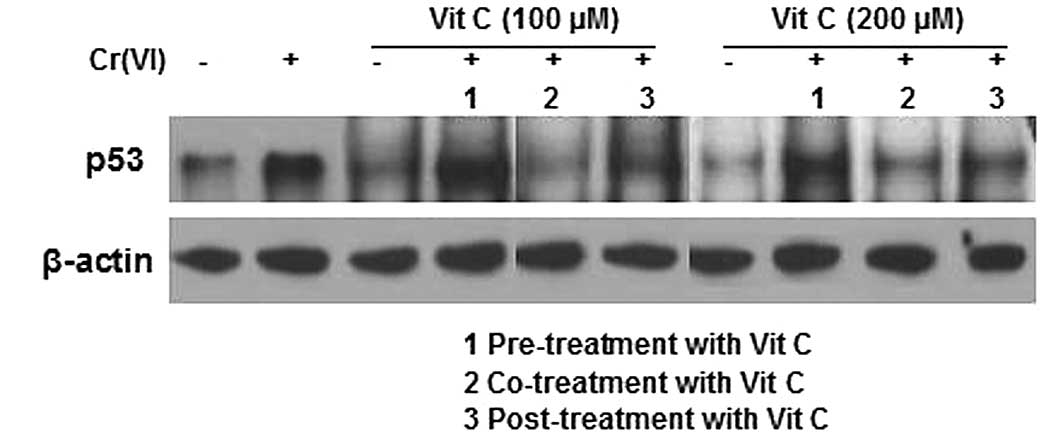
p53 expression correlates with the
Cr(VI)-induced increase in DPC formation
A previous study suggested a possible correlation
between p53 expression and DPC formation following formaldehyde
exposure (31). Thus, we detected
p53 expression levels using western blot analysis. Cr(VI) induced
higher p53 expression levels. The p53 expression levels of the
treatment groups receiving vit C alone (100 and 200 μM) were
slightly decreased when compared with that of the control group.
Compared with the treatment group receiving Cr(VI) alone, the p53
levels in the vit C pre-treatment plus Cr(VI) groups were
significantly increased. Additionally, while the Cr(VI) plus vit C
post-treatment groups revealed no significant change in p53 levels
compared with the treatment groups receiving vit C alone, the
Cr(VI) and vit C co-treatment groups demonstrated a significant
decrease in p53 levels (Fig. 5).
The changes in p53 expression in the groups of different time-order
treatments of vit C with Cr(VI) corresponded with the changes in
the DPC coefficient (Fig. 2B);
thus, we concluded that p53 expression was correlated with the
Cr(VI)-induced increase of DPC.
Discussion
Numerous studies have demonstrated that Cr(VI) is
genotoxic and mutagenic to mammalian cells. Cr(VI) is transported
into cells through the sulfate anion transport system (32), and is immediately reduced by the
redox system to its stable form, Cr(III), once inside cells
(33). It has been suggested that
the ROS that are generated during the cellular reduction process
may initiate the carcinogenic process by altering DNA structures
(16). Various chemical and
physical agents, which are known or suspected carcinogens, such as
Cr(VI), are able to induce DPC formation. One of the most commonly
occurring types of DNA damage following Cr(VI) exposure is DPC
formation (29). DPCs are large
helix-distorting lesions that interfere with DNA replication,
transcription, repair and recombination, as well as with chromatin
remodeling (34). By blocking the
binding and progression of protein complexes, DPC formation results
in severe cytotoxic and mutagenic outcomes (35,36).
DPC levels are used as biomarkers for detecting Cr(VI)-associated
genetic damage in the exposed individuals, as they are not
significantly affected by age, gender and body weight (37). Experimental studies have suggested
that Cr(VI) causes DPC formation either by Cr(III)-associated
cross-linking reactions or by oxidative mechanisms (38,39).
However, to what extent the DPCs are directly responsible for the
formation of mutations and cancer remains unclear.
Mitochondria are well known for their functions of
supplying cellular energy and signaling, as well as regulating the
cell cycle and cell differentiation. Mitochondrial damage may lead
to severe consequences, including cell death (40,41),
and it is important in Cr(VI)-induced cytotoxicity (42). The PTP is an inner membrane
megachannel, the opening of which results in mitochondrial
swelling, depolarization and rupture of the outer membrane
(43). The decrease in MMP leads
to matrix condensation and the exposure of cytochrome c to the
intermembrane space, which is involved in mediating mitochondrial
dysfunction (44). The MDA content
may be viewed as a biomarker of mitochondrial oxidative stress, as
MDA may cause cellular toxic stress and form covalent protein
adducts to interrupt the normal function of mitochondria (45). In the present study, we observed
that Cr(VI) induced mitochondrial damage, which was characterized
by the increased MDA content and open PTP rate, as well as the
decreased MMP. In addition, we identified that ROS levels were
correlated with mitochondrial damage.
ROS, including O2• − and
H2O2, are the products of normal cellular
functioning; however, excessive ROS levels may also cause
deleterious effects. Following Cr(VI) exposure, ROS were observed
to be generated not only from the reduction process mediated by the
redox system; however, also from the electrons leaked from the
electron transport chain (ETC), when mitochondrial respiratory
chain complexes (MRCC) were inhibited by Cr(VI) (46,47).
ROS levels are correlated with mitochondrial damage, as
mitochondrial dysfunction, including oxidative stress, results in
ROS accumulation. This accumulation may aggravate mitochondrial
damage by attacking mitochondrial DNA, deactivating specific
enzymes and inhibiting the function of the ETC. By determining the
time-order effect of the small molecule antioxidant, vit C, on
Cr(VI)-induced mitochondrial damage in PBLs, we concluded that vit
C pre-treatment and co-treatment had a protective effect against
Cr(VI)-induced mitochondrial damage and loss of cell viability.
This was due to the fact that vit C inhibited cellular ROS by
suppressing ROS production and scavenging excessive accumulated
ROS. It has been demonstrated that ROS accumulation occurred within
only a few minutes following Cr(VI) exposure, thus the
2-h-postponed treatment of vit C was too late to clear the ROS
accumulation and to rescue the ROS-induced damage. Therefore, this
explains why, in our study, the Cr(VI) plus vit C post-treatment
groups did not exhibit any protective effects against mitochondrial
damage and loss of cell viability.
The tumor suppressor, p53, was identified in 1979,
and it is well known that p53 activation is involved in growth
arrest and apoptosis. It was demonstrated that high p53 serum
levels were identifiable several years prior to the cancer was able
to be clinically detected (48).
In a previous study by Hanaoka et al, high levels of serum
pantropic p53 proteins were detected in workers that had a long
history of Cr(VI) exposure, and thus were presumed to be at high
risk for lung cancer (49). In the
present study, we concluded that p53 expression was associated with
the Cr(VI)-induced increase in DPC formation. We observed that when
PBLs were exposed to Cr(VI) together with different time-orders of
vit C treatments, only vit C co-treatment demonstrated a protective
effect against the Cr(VI)-induced increase in DPCs. Cr(VI) may be
reduced outside the cell by vit C to Cr(III), which cannot cross
cell membranes due to its tendency to form insoluble hydrated
complexes; this explains why the lowest number of DPCs occurred in
the vit C co-treatment groups. The reason for the increased
formation of DPCs in the vit C pre-treatment plus Cr(VI) group is
that vit C pre-treatment enhanced the capacity of the cell to
reduce Cr(VI), and the accumulation of Cr(III) resulted in the
formation of Cr(III)-DNA binding adducts. This is consistent with a
previous study conducted by Mattagajasingh et al(16). The Cr(VI) plus vit C post-treatment
groups revealed no significant changes in DPC generation compared
with the Cr(VI) alone group, as the postponed treatment with vit C
was too late to halt the toxicity of Cr(VI).
Cr(III) is proposed to give rise to DPCs by directly
mediating the cross linking between cellular DNA and proteins
(50). A previous study provided
evidence for a three-step cross-linking mechanism, which included
reduction of Cr(VI) to Cr(III), Cr(III)-DNA binding and the
generation of protein-Cr(III)-DNA crosslinks (51). Shaham et al demonstrated a
possible causal correlation between p53 activation and DPC
formation following the examination of the PBLs of 186 workers
exposed to formaldehyde (31). The
authors also suggested that p53 activation and DPC formation may
represent steps in formaldehyde carcinogenesis. The mechanism
whereby p53 activation correlates with DPC formation following
Cr(VI) exposure in PBLs remains unclear. Previous studies have
demonstrated that ROS are able to increase DPC levels by the
initial oxidative lesions on DNA, as well as by the reactive
products of lipid peroxidation, including MDA (52). In the present study, we did not
observe a correlation between ROS levels and the formation of DPCs.
Cr(VI) and its compound-induced gene mutations have been
demonstrated in a variety of cell systems (53,54),
and it is suggested that the induction of DPC formation is
associated with chromosomal effects; however not with the
occurrence of gene mutations (55). Thus, we inferred that
Cr(VI)-induced DPCs were not the cause of gene mutations involved
in carcinogenesis, and that the mutations of tumor-related genes
following exposure to Cr(VI) are not a direct result of DPCs.
We concluded that vit C has different time-order
effects on Cr(VI)-induced mitochondrial damage and DPC formation.
Although further investigation is required, it is clear that the
biomarkers, including DPC and p53, may be used in the assessment of
the development of Cr(VI)-induced cancers. Therefore, this
facilitates a closer follow-up of populations exposed to Cr(VI),
for secondary prevention.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (Grant no. 81172701) and Open-End Fund
for the Valuable and Precision Instruments of Central South
University (No. CSUZC2013047).
Abbreviations:
|
PTP
|
permeability transition pore
|
|
MMP
|
mitochondrial membrane potential
|
|
DPC
|
DNA-protein crosslink
|
|
ROS
|
reactive oxygen species
|
|
PBLs
|
peripheral blood lymphocytes
|
|
MTT
|
3-(4,5-dimethylthiazol-2yl-)-2,5-diphenyl tetrazolium bromide
|
|
ETC
|
electron transport chain
|
|
MRCC
|
mitochondrial respiratory chain
complex
|
References
|
1
|
Nriagu JO and Nieboer E: Chromium in the
Natural and Human Environments. 1st edition. Wiley; New York:
1988
|
|
2
|
Katz SA and Salem H: The Biological and
Environmental Chemistry of Chromium. VCH Publishers; New York:
1994
|
|
3
|
Voitkun V, Zhitkovich A and Costa M:
Cr(III)-mediated crosslinks of glutathione or amino acids to the
DNA phosphate backbone are mutagenic in human cells. Nucleic Acids
Res. 26:2024–2030. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sathwara NG, Patel KG, Vyas JB, et al:
Chromium exposure study in chemical based industry. J Environ Biol.
28:405–408. 2007.PubMed/NCBI
|
|
5
|
Chun L, Hongzhang C and Zuohu L:
Adsorptive removal of Cr(VI) by Fe-modified steam exploded wheat
straw. Process Biochem. 39:541–545. 2004. View Article : Google Scholar
|
|
6
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: more than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hodges NJ, Adám B, Lee AJ, Cross HJ and
Chipman JK: Induction of DNA-strand breaks in human peripheral
blood lymphocytes and A549 lung cells by sodium dichromate:
association with 8-oxo-2-deoxyguanosine formation and
inter-individual variability. Mutagenesis. 16:467–474. 2001.
View Article : Google Scholar
|
|
8
|
Costa M, Zhitkovich A and Toniolo P:
DNA-protein cross-links in welders: molecular implications. Cancer
Res. 53:460–463. 1993.PubMed/NCBI
|
|
9
|
Budhwar R, Das M, Bihari V and Kumar S:
Exposure estimates of chromeplaters in India: an exploratory study.
Biomarkers. 10:252–257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Medeiros MG, Rodrigues AS, Batoréu MC,
Laires A, Rueff J and Zhitkovich A: Elevated levels of DNA-protein
crosslinks and micronuclei in peripheral lymphocytes of tannery
workers exposed to trivalent chromium. Mutagenesis. 18:19–24. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhitkovich A, Lukanova A, Popov T, et al:
DNA-protein crosslinks in peripheral lymphocytes of individuals
exposed to hexavalent chromium compounds. Biomarkers. 1:86–93.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Quievryn G, Peterson E, Messer J and
Zhitkovich A: Genotoxicity and mutagenicity of
chromium(VI)/ascorbate-generated DNA adducts in human and bacterial
cells. Biochemistry. 42:1062–1070. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
De Flora S and Wetterhahn KE: Mechanisms
of chromium metabolism and genotoxicity. Life Chem Rep. 7:169–244.
1989.
|
|
14
|
Wei YD, Tepperman K, Huang M, Sartor MA
and Puga A: Chromium inhibits transcription from polycyclic
aromatic hydrocarbon-inducible promoters by blocking the release of
histone deacetylase and preventing the binding of p300 to
chromatin. J Biol Chem. 279:4110–4119. 2004. View Article : Google Scholar
|
|
15
|
Schnekenburger M, Talaska G and Puga A:
Chromium cross-links histone deacetylase 1-DNA methyltransferase 1
complexes to chromatin, inhibiting histone-remodeling marks
critical for transcriptional activation. Mol Cell Biol.
27:7089–7101. 2007. View Article : Google Scholar
|
|
16
|
Mattagajasingh SN and Misra HP: Mechanisms
of the carcinogenic chromium(VI)-induced DNA-protein cross-linking
and their characterization in cultured intact human cells. J Biol
Chem. 271:33550–33560. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ali SF, LeBel CP and Bondy SC: Reactive
oxygen species formation as a biomarker of methylmercury and
trimethyltin neurotoxicity. Neurotoxicology. 13:637–648.
1992.PubMed/NCBI
|
|
18
|
Valko M, Rhodes CJ, Moncol J, Izakovic M
and Mazur M: Free radicals, metals and antioxidants in oxidative
stress-induced cancer. Chem Biol Interact. 160:1–40. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu XR, Li HB, Gu JD and Li XY: Kinetics of
the reduction of chromium(VI) by vitamin C. Environ Toxicol Chem.
24:1310–1314. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chorvatovicová D, Ginter E, Kosinová A and
Zloch Z: Effect of vitamins C and E on toxicity and mutagenicity of
hexavalent chromium in rat and guinea pig. Mutat Res. 262:41–46.
1991.PubMed/NCBI
|
|
21
|
Stearns DM, Kennedy LJ, Courtney KD,
Giangrande PH, Phieffer LS and Wetterhahn KE: Reduction of
chromium(VI) by ascorbate leads to chromium-DNA binding and DNA
strand breaks in vitro. Biochemistry. 34:910–919. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szablewski L, Sobczyk-Kopcioł A, Oleszczak
B, Mrozikiewicz-Rakowska B, Karnafel W and Grytner-Zięcina B:
Expression of glucose transporters in peripheral blood cells in
patients with type 2 diabetes mellitus depending on the mode of
therapy. Diabetologia Doświadczalna i Kliniczna. 7:204–212.
2007.
|
|
23
|
Piątkiewicz P, Czech A, Tatoń J and Górski
A: Investigations of cellular glucose transport and its regulation
under the influence of insulin in human peripheral blood
lymphocytes. Endokrynol Pol. 61:182–187. 2010.PubMed/NCBI
|
|
24
|
Cinatl J Jr, Cinatl J, Driever PH, et al:
Sodium valproate inhibits in vivo growth of human neuroblastoma
cells. Anticancer Drugs. 8:958–963. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhitkovich A and Costa M: A simple,
sensitive assay to detect DNA-protein crosslinks in intact cells
and in vivo. Carcinogenesis. 13:1485–1489. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brustovetsky N, Brustovetsky T, Jemmerson
R and Dubinsky JM: Calcium-induced cytochrome c release from CNS
mitochondria is associated with the permeability transition and
rupture of the outer membrane. J Neurochem. 80:207–218. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Emaus RK, Grunwald R and Lemasters JJ:
Rhodamine 123 as a probe of transmembrane potential in isolated
rat-liver mitochondria: spectral and metabolic properties. Biochim
Biophys Acta. 850:436–448. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhitkovich A, Voitkun V, Kluz T and Costa
M: Utilization of DNA-protein cross-links as a biomarker of
chromium exposure. Environ Health Perspect. 106(Suppl 4): 969–974.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li CJ, Zhang QM, Li MZ, Zhang JY, Yu P and
Yu DM: Attenuation of myocardial apoptosis by alpha-lipoic acid
through suppression of mitochondrial oxidative stress to reduce
diabetic cardiomyopathy. Chin Med J (Engl). 122:2580–2586.
2009.PubMed/NCBI
|
|
31
|
Shaham J, Bomstein Y, Gurvich R,
Rashkovsky M and Kaufman Z: DNA-protein crosslinks and p53 protein
expression in relation to occupational exposure to formaldehyde.
Occup Environ Med. 60:403–409. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jennette KW: The role of metals in
carcinogenesis: biochemistry and metabolism. Environ Health
Perspect. 40:233–252. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Connett PH and Wetterhahn KE: Metabolism
of the carcinogen chromate by cellular constituents. Struct
Bonding. 54:93–124. 1983. View Article : Google Scholar
|
|
34
|
Michaelson-Richie ED, Loeber RL, Codreanu
SG, et al: DNA-protein cross-linking by 1,2,3,4-diepoxybutane. J
Proteome Res. 9:4356–4357. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Barker S, Weinfeld M and Murray D:
DNA-protein crosslinks: their induction, repair, and biological
consequences. Mutat Res. 589:111–135. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Oleinick NL, Chiu SM, Ramakrishnan N and
Xue LY: The formation, identification, and significance of
DNA-protein cross-links in mammalian cells. Br J Cancer. (Suppl 8):
135–140. 1987.PubMed/NCBI
|
|
37
|
Taioli E, Zhitkovich A, Toniolo P and
Costa M: Normal values of DNA-protein crosslinks in mononuclear
cells of a population of healthy controls. Cancer J. 8:76–79.
1995.
|
|
38
|
Xu X, Muller JG, Ye Y and Burrows CJ:
DNA-protein cross-links between guanine and lysine depend on the
mechanism of oxidation for formation of C5 vs C8 guanosine adducts.
J Am Chem Soc. 130:703–709. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bjorklund CC and Davis WB: Stable
DNA-protein cross-links are products of DNA charge transport in a
nucleosome core particle. Biochemistry. 46:10745–10755. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Giorgi C, Romagnoli A, Pinton P and
Rizzuto R: Ca2+ signaling, mitochondria and cell death.
Curr Mol Med. 8:119–130. 2008.
|
|
41
|
Nisoli E, Clementi E, Moncada S and
Carruba MO: Mitochondrial biogenesis as a cellular signaling
framework. Biochem Pharmacol. 67:1–15. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Son YO, Hitron JA, Wang X, et al: Cr(VI)
induces mitochondrial-mediated and caspase-dependent apoptosis
through reactive oxygen species-mediated p53 activation in JB6 CI41
cells. Toxicol Appl Pharmacol. 245:226–235. 2010. View Article : Google Scholar
|
|
43
|
Szabó I and Zoratti M: The mitochondrial
megachannel is the permeability transition pore. J Bioenerg
Biomembr. 24:111–117. 1992.PubMed/NCBI
|
|
44
|
Gottlieb E, Armour SM, Harris MH and
Thompson CB: Mitochondrial membrane potential regulates matrix
configuration and cytochrome c release during apoptosis. Cell Death
Differ. 10:709–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tuma DJ, Thiele GM, Xu D, Klassen LW and
Sorrell MF: Acetaldehyde and malondialdehyde react together to
generate distinct protein adducts in the liver during long-term
ethanol administration. Hepatology. 23:872–880. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xiao F, Feng X, Zeng M, Guan L, Hu Q and
Zhong C: Hexavalent chromium induces energy metabolism disturbance
and p53-dependent cell cycle arrest via reactive oxygen species in
L-02 hepatocytes. Mol Cell Biochem. 371:65–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xiao F, Li Y, Dai L, et al: Hexavalent
chromium targets mitochondrial respiratory chain complex I to
induce reactive oxygen species-dependent caspase-3 activation in
L-02 hepatocytes. Int J Mol Med. 30:629–635. 2012.
|
|
48
|
Hemminki K, Partanen R, Koskinen H, Smith
S, Carney W and Brandt-Rauf PW: The molecular epidemiology of
oncoproteins. Serum p53 protein in patients with asbestosis. Chest.
109(3 Suppl): 22S–26S. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hanaoka T, Yamano Y, Katsuno N, Kagawa J
and Ishizu S: Elevated serum levels of pantropic p53 proteins in
chromium workers. Scand J Work Environ Health. 23:37–40. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kortenkamp A, Curran B and O’Brien P:
Defining conditions for the efficient in vitro cross-linking of
proteins to DNA by chromium(III) compounds. Carcinogenesis.
13:307–308. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Macfie A, Hagan E and Zhitkovich A:
Mechanism of DNA-protein cross-linking by chromium. Chem Res
Toxicol. 23:341–347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Voitkun V and Zhitkovich A: Analysis of
DNA-protein crosslinking activity of malondialdehyde in vitro.
Mutat Res. 424:97–106. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Singh J, Carlisle DL, Pritchard DE and
Patierno SR: Chromium-induced genotoxicity and apoptosis:
relationship to chromium carcinogenesis (review). Oncol Rep.
5:1307–1318. 1998.PubMed/NCBI
|
|
54
|
Bianchi V, Celotti L, Lanfranchi G, et al:
Genetic effects of chromium compounds. Mutat Res. 117:279–300.
1983. View Article : Google Scholar
|
|
55
|
Merk O and Speit G: Significance of
formaldehyde-induced DNA-protein crosslinks for mutagenesis.
Environ Mol Mutagen. 32:260–268. 1998. View Article : Google Scholar : PubMed/NCBI
|