Introduction
Hepatic encephalopathy (HE) is a clinical
neuropsychiatric syndrome resulting from a metabolic disturbance
caused by severe liver disease and/or portal-systemic shunts. HE is
associated with significant morbidity and mortality (1). Several theories have been proposed to
explain the pathogenesis of HE. One such theory is the ammonia
hypothesis, which states that the level of ammonia plays a central
role in the pathogenesis of HE. Infection, inflammation, oxidative
stress and steroid hormones are also considered to be important
(2). As the precise underlying
mechanism of the pathogenesis of HE remains unclear and specific
treatments for HE are limited, the reduction of ammonia levels is
regarded as the primary approach to prevent HE. Gaining a better
understanding of the pathogenesis of HE is important for the
development of effective treatment strategies in the future.
Heme oxygenase-1 (HO-1) catalyzes the oxidative
degradation of heme to carbon monoxide (CO), free iron and
biliverdin (3). In normal brain
tissue, basal HO-1 expression levels and activity are maintained at
low levels and are confined to the neuroglia and occasionally, the
scattered neurons (4). However,
the level of HO-1 activity may be significantly enhanced in
astrocytes, microglia and certain neurons by extravascular
hemoglobin, hemin and oxidants (5,6). The
upregulation of HO-1 attenuates post-ischemic brain damage via
simultaneous inhibition of superoxide production and preservation
of nitric oxide (NO) bioavailability (7). It has been reported that HO-1 may be
a therapeutic target in neurodegenerative disease and brain
infection (8).
Our previous studies have indicated that HO-1
expression is upregulated in the liver and the HO/CO pathway may
regulate cirrhosis induced in rats by bile duct ligation (BDL), as
well as its complications, including hepatorenal and
hepatopulmonary syndrome (9,10).
In addition, the levels of carboxyhemoglobin (COHb) in the arterial
blood of hepatitis B virus-related cirrhosis patients with HE were
significantly increased compared with healthy individuals (11). The mechanism by which HO-1 is
expressed in the brain and affects the development and progression
of HE remains unclear.
The present study aimed to evaluate whether the
regulation of HO-1 affects the development and progression of HE,
and the mechanism by which its products affect cerebral
metabolism.
Materials and methods
Animal care
The experimental protocols used in the present study
were approved by the Animal Care and Use Committee of Dalian
Medical University (Liaoning, China) in accordance with the
guidelines established by the China Council on Animal Care.
Generation of HE models and the treatment
of rats
The 46 healthy male Sprague Dawley rats, weighing
200–220 g, were obtained from the Laboratory Animal Center of
Dalian Medical University (Dalian, China) and randomly divided into
5 treatment groups; sham (n=6), BDL (n=10), HE (n=12), zinc
protoporphyrin (ZnPP; n=8) and cobalt protoporphyrin (CoPP; n=10).
The rats were housed in a specific pathogen-free center at room
temperature (24–26°C) and a relative humidity of 60–65%. Water was
provided ad libitum.
The rats were fed and housed for 3 days prior to any
experimental protocols being conducted. All surgical procedures
were approved by the Animal Care and Use Committee of Dalian
Medical University. Laparotomy was performed under anesthesia with
ether. The common bile duct was localized, doubly ligated and cut
between these two ligatures. In the sham group, a midline incision
was performed in the animals; however, this was without BDL. Four
groups underwent BDL and a sham surgery group was used as a
control. Two weeks following surgery, the sham and BDL rats were
pair-fed and administered with an i.p. saline injection. The HE,
ZnPP and CoPP groups were fed on an ammonium-containing diet
(ammonium acetate, 20% w/w), as described previously (12), and were administered with an i.p.
saline, ZnPP or CoPP injection (5 mg/kg body weight), three times
per week, respectively. The animals were treated with ZnPP and CoPP
in order to downregulate and upregulate the expression of HO-1,
respectively. Following the establishment of the HE models, the
number of rats in each group was reduced to 6 due to deaths
encountered within specific groups.
ZnPP and CoPP (Sigma, St. Louis, MO, USA) were
dissolved in 0.2 mol/l NaOH, adjusted to pH 7.4 and diluted in
0.85% NaCl to 1 mg/ml, as described previously (13). Histostain™-Plus (SP9001; Zhongshan
Goldenbridge Biological Technology, Beijing, China), superoxide
dismutase (SOD) and malonaldehyde (MDA) kits (Nanjing KeyGen
Biotech. Co., Ltd., Nanjing, China) were used in the study.
Sample collection and examination
Two weeks following treatment, a catheter connected
to a Pressure Transducer (BL-420F biological experimental system;
Chengdu Technology and Market Co., Ltd., Chengdu, China) was placed
in the portal vein to measure portal vein pressure (PVP).
Subsequently, 1 ml of arterial blood was withdrawn to measure serum
COHb levels using a Rapid Lab 1245 Blood Gas Analyzer (Siemens, New
York, NY, USA). Levels of alanine aminotransferase (ALT), aspartate
aminotransferase (AST), total bilirubin (TBIL) and serum iron were
detected using a Hitachi 7600-110 Automatic Biochemical Analyzer
(Hitachi Co., Tokyo, Japan).
Measurement of plasma and cerebral
ammonia levels
The levels of ammonia were measured in plasma and
the cerebral cortex. Blood samples were examined using the Ammonia
Checker II (KEM, Kyoto, Japan). The cerebral ammonia levels were
measured by fluorimetry (Fluoroskan Ascent Labsystems, Helsinki,
Finland), according to the methods described previously (12). Assays were performed in Costar
96-well UV plates (Corning Costar Corporation, Cambridge, MA,
USA).
Measurement of brain water content
The levels of brain water were quantitated by the
wet-weight/dry-weight method. Half of the brain was weighed prior
to and following 48 h incubation in a 120°C oven. The brain water
content percentage was calculated using the following formula:
Brain water content (%) = (wet weight − dry weight)/wet weight ×
100%, as described previously (14).
Measurement of locomotor activity
Locomotor activity was assessed using an infrared
beam computerized auto-track system (Columbus Instruments,
Columbus, OH, USA) (15). Prior to
being sacrificed, rats from each of the 5 groups were individually
placed in plexiglass cages (29×22×22 cm) for 6 h before activity
was recorded. Cumulative distance traveled during the day (inactive
period) and night (active period) was recorded for 24 h and
expressed as the night/day ratio.
Measurement of oxidative stress
Levels of brain SOD and MDA were determined using
the UV-2100 Spectrophotometer (Chemito Instruments Pvt. Ltd.,
Mumbai, India), according to the manufacturer’s instructions.
Histology and immunohistochemistry
Sections of the liver and brain lobe were excised,
fixed in 10% neutral formalin solution and embedded in paraffin.
Hematoxylin and eosin (H&E) staining was performed according to
standard procedure. Lesion severity was graded as described
previously (16). Tissue sections
(4-μm thick) were briefly treated with HCl (5%) to liberate ferric
ions. Samples were then treated with 5% potassium ferrocyanide to
produce insoluble ferric ferrocyanide. Slides were counterstained
with neutral red. For immunohistochemical examination,
deparaffinized sections were incubated with HO-1 antibodies (Abcam,
Cambridge, MA, USA; 1:1,000 dilution), aquaporin-4 (AQP-4)
antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA;
1:400 dilution) and biotinylated secondary antibodies (Santa Cruz
Biotechnology, Inc.), followed by avidin-biotin-peroxidase complex.
Yellow material in the cytoplasm was considered to represent a
positive cell. Cell staining was assigned to 4 scores, as described
previously (17). The final score
was defined as follows: staining intensity × percentage of positive
cells. The mean score of 5 fields was used to compare the 5
groups.
Western blot analysis
The resected liver tissues were extracted with lysis
buffer (1% Triton X-100, 50 mmol/l Tris-HCl pH 7.6, 150 mmol/l NaCl
and 1% protease inhibitor cocktail). Western blotting was performed
in accordance with a previously described protocol (18). Western blot analysis was performed
with liver homogenates (30 μg protein) using anti-HO-1 antibody
(Abcam; 1:2,000 dilution), anti-β-actin antibody (Zhongshan
Goldenbridge Biological Technology; 1:500 dilution) and secondary
anti-rabbit and anti-mouse IgG (Santa Cruz Biotechnology, Inc.;
1:500 dilution). The intensity of each signal was corrected by the
values obtained from the immunodetection of β-actin and the
relative protein intensity was expressed as the fold-change of the
content in the control group.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was used to analyze the expression levels of
HO-1 and AQP-4. Total RNA was extracted from each chamber using
RNAiso Plus ((Takara Bio, Inc., Dalian, China). The total RNA was
then reverse transcribed into cDNA using the
PrimeScript® RT reagent kit (Perfect Real Time; Takara
Bio, Inc.). Real-time RT-PCR was performed with a Mx3000P QPCR
System (Agilent Technologies, Palo Alto, CA, USA) using
SYBR® Premix Ex Taq II (Perfect Real Time). β-Actin, a
common housekeeping gene in cells, was used as the internal control
gene to normalize the quantities of target gene expression.
Thermocycling conditions were as follows: 95°C for 30 sec, 40
cycles of denaturation (95°C, 5 sec), annealing (60°C, 30 sec) and
extension (72°C, 30 sec). The primers used in the RT-PCR are listed
in Table I.
 | Table IPrimers used for real-time PCR
analysis |
Table I
Primers used for real-time PCR
analysis
| Gene |
Forward/reverse | Sequence 5′-3′ |
|---|
| HO-1 | Forward |
AGGTGCACATCCGTGCAGAG |
| Reverse |
CTTCCAGGGCCGTATAGATATGGTA |
| AQP-4 | Forward |
TTGGACCAATCATAGGCGC |
| Reverse |
GGTCAATGTCGATCACATGC |
| β-actin | Forward |
GGAGATTACTGCCCTGGCTCCTA |
| Reverse |
GACTCATCGTACTCCTGCTTGCTG |
Statistical analysis
All data are presented as the mean ± SD. Statistical
analysis was performed using SPSS software version 16.0 (IBM,
Chicago, IL, USA). Groups were compared using one-way ANOVA with
the Dunnett’s multiple comparison test (where applicable).
P<0.05 was considered to indicate a statistically significant
result.
Results
Diet-induced HE model in BDL rats
Common BDL, ascites and jaundice were observed in
BDL rats at 4 weeks post-surgery. The serum levels of AST, ALT and
TBIL in the BDL group were significantly higher compared with the
sham group (P<0.01). In addition, the serum levels of AST, ALT
and TBIL were significantly increased in the HE group compared with
the BDL group (P<0.01; Fig.
1B).
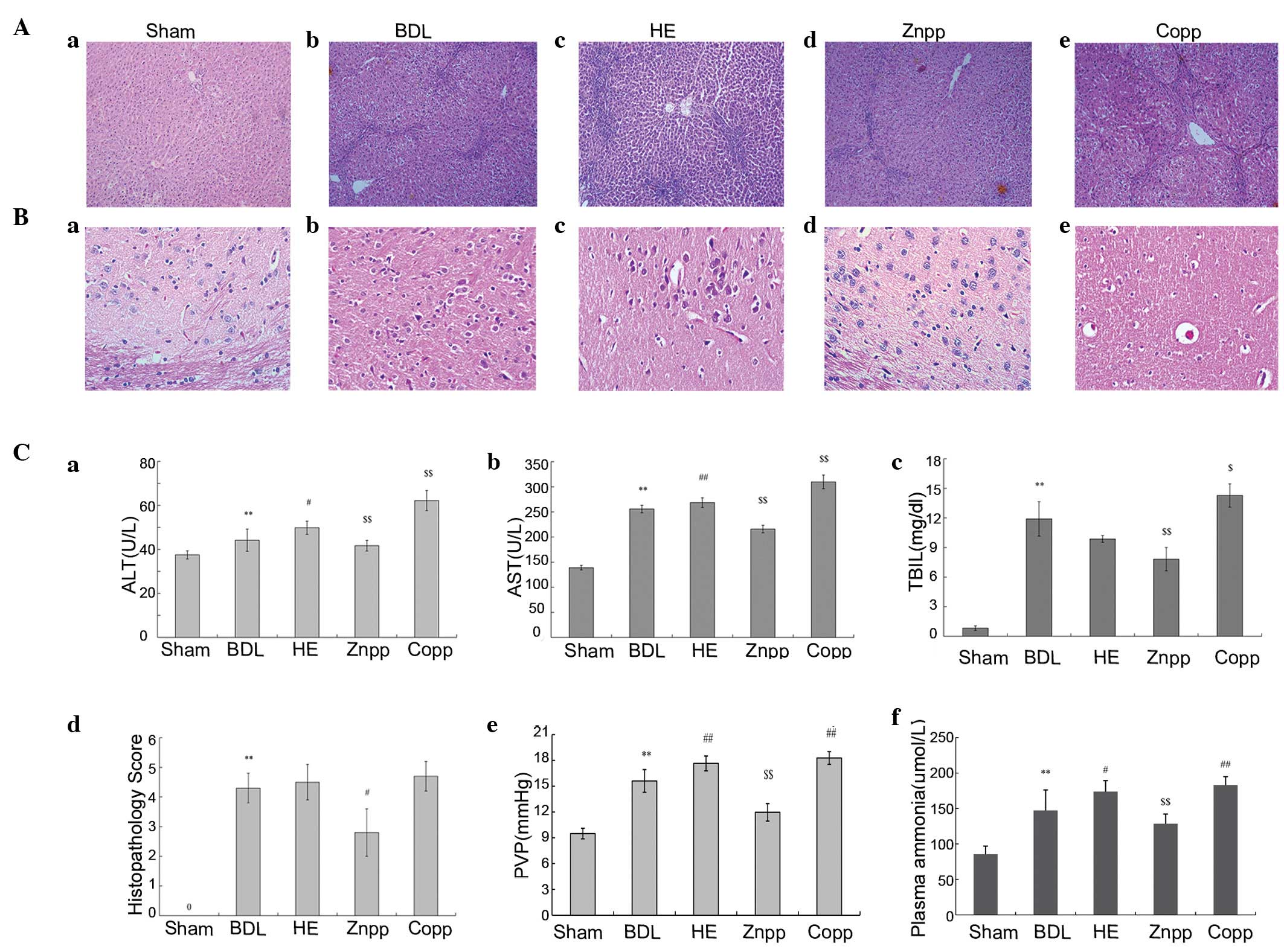 | Figure 1Assessment of HE. (A and B) Liver and
brain H&E staining. (A) Normal lobular architecture observed in
the (Aa) sham group; fibrous hyperplasia in the (Ab) BDL, (Ac) HE,
(Ad) ZnPP and (Ae) CoPP groups. Less severe fibrous hyperplasia was
observed in the ZnPP group compared with the BDL group (original
magnification, ×100). (Ba) The brain tissue of the sham group
demonstrated a normal cellularity and architecture; the cerebral
intracellular edema of the brain in the (Bb) BDL, (Bc) HE, (Bd)
ZnPP and (Be) CoPP groups. The cerebral intracellular edema was
more severe in the HE and CoPP groups compared with the BDL group;
however, it was mitigated in the ZnPP group (original
magnification, ×400). (C) Serum index and PVP; (Ca) ALT, (Cb) AST
and (Cc) TBIL levels were significantly increased in the HE and
CoPP groups compared with the BDL group; however, they were
significantly decreased in the ZnPP group. In addition, the AST,
ALT and TBIL levels were lower in the ZnPP group compared with the
HE group; (Cd) hepatic fibrosis was assessed using
histopathological scoring; (Ce) the PVP was higher in the BDL group
compared with the sham group; however, was significantly increased
in the HE and CoPP groups compared with the BDL group, and lower in
the ZnPP group; (Cf) levels of plasma ammonia were higher in the HE
and CoPP groups compared with the BDL group, and lower in the ZnPP
group. The data are presented as the mean ± SD.
**P<0.01, *P<0.05 vs. sham;
##P<0.01, #P<0.05 vs. BDL;
$$P<0.01, $P<0.05 vs. HE. H&E,
hematoxylin and eosin; HE, hepatic encephalopathy; AST, aspartate
aminotransferase; ALT, alanine aminotransferase; TBIL, total
bilirubin; BDL, bile duct ligation; CoPP, cobalt protoporphyrin;
ZnPP, zinc protoporphyrin; PVP, portal vein pressure. |
PVP was significantly higher in the BDL group
compared with the sham group (P<0.01). Compared with the BDL
group, the PVP was significantly elevated in HE rats. In addition,
PVP decreased following the treatment with ZnPP and increased
following the treatment with CoPP compared with HE rats (P<0.01;
Fig. 1Be). Plasma ammonia levels
were higher in the BDL group compared with the sham controls.
Higher plasma ammonia levels were observed in the HE group compared
with the BDL group (P<0.01; Fig.
1Cf).
Hepatic fibrosis was evaluated by H&E staining.
Compared with the sham group, the BDL and HE groups exhibited
increased levels of inflammation, necrosis and destruction of the
lobular architecture (Fig. 1A).
Brain H&E staining demonstrated normal cellularity and
architecture in the sham group. Compared with the BDL group, HE
rats demonstrated more severe cerebral intracellular edema in the
brain (Fig. 1B).
Inhibition of HO-1 expression improves
HE
The HO-1 mRNA and protein expression levels in the
brain increased significantly following BDL treatment compared with
the sham controls, and were significantly elevated in the HE group
compared with the BDL group (P<0.01). In addition, the mRNA and
protein expression levels of HO-1 were significantly decreased in
the ZnPP treatment group compared with the HE group; however, these
were enhanced in the CoPP treatment group (Fig. 2B). The COHb levels in the arterial
blood were in accordance with HO-1 expression (Fig. 2Ca). Cerebral HO-1 immunostaining
demonstrated that HO-1 was mainly expressed in the cerebral cortex
(Fig. 2A).
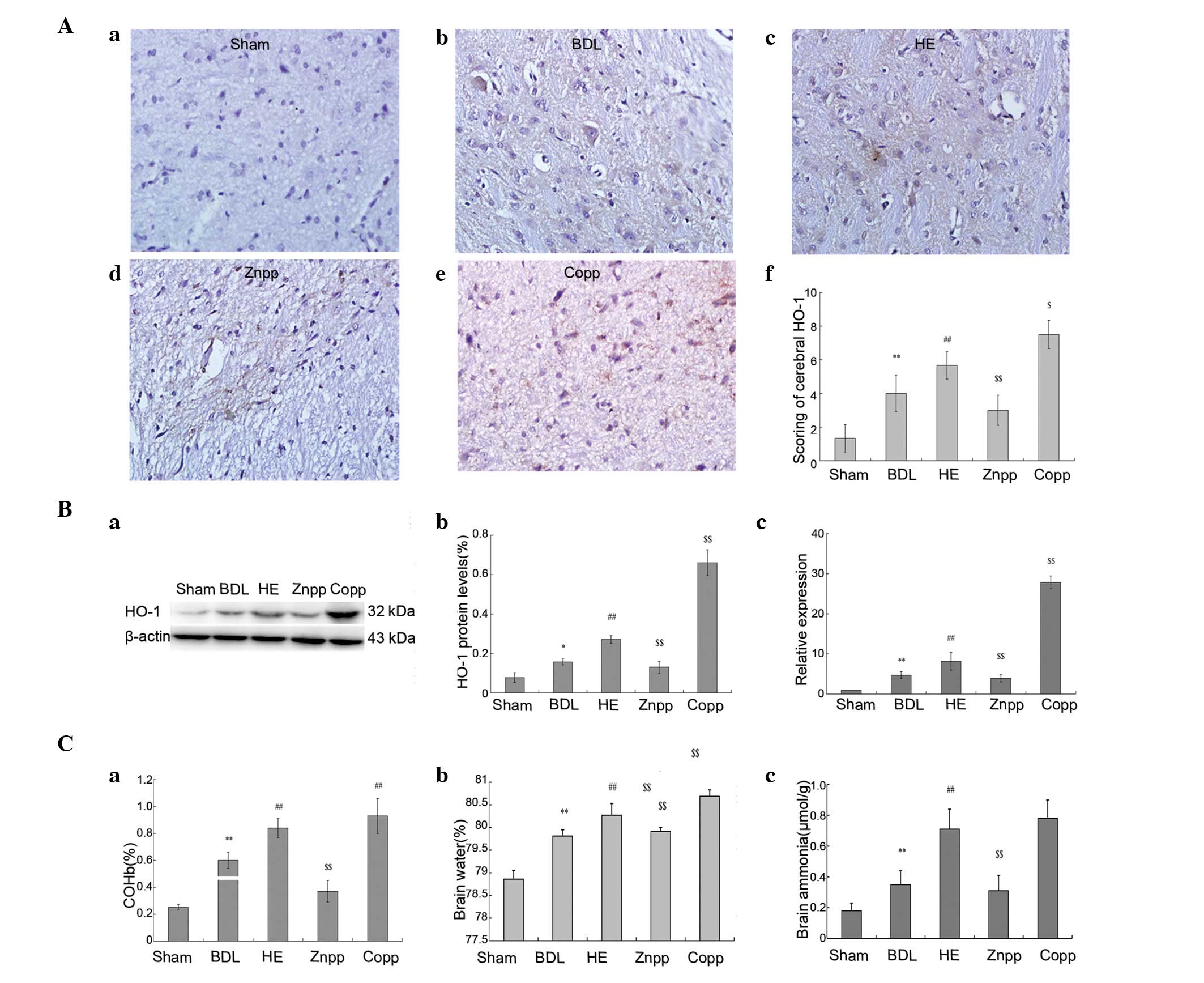 | Figure 2Levels of HO-1, ammonia and brain
water content. (A) Immunohistochemical staining with HO-1; (Aa)
minimal levels of HO-1 expression were detected in the cerebral
cortex in the sham group; HO-1 expression was observed in the (Ab)
BDL, (Ac) HE, (Ad) ZnPP and (Ae) CoPP groups. HO-1 was seldomly
expressed in the ZnPP group; (Af) quantitative scoring of the
immunohistochemical staining of cerebral HO-1 protein expression
(original magnification, ×400). (B) Protein and mRNA expression
levels of HO-1; (Ba and Bb) HO-1 protein expression was higher in
the HE and CoPP groups and decreased in the ZnPP group compared
with the BDL group; (Bc) HO-1 mRNA levels were quantified using
real-time PCR in the brain tissue. (C) Levels of COHb, ammonia and
water content. Serum COHb levels in the arterial blood were
quantified. (Ca) Higher levels of COHb were observed in the HE and
CoPP groups compared with the BDL group, and were lower in the ZnPP
group; (Cb) brain water content and (Cc) ammonia levels in the HE
and CoPP groups were enhanced compared with the BDL group, and
decreased in the ZnPP group. The data are presented as the mean ±
SD. **P<0.01, *P<0.05 vs. sham;
##P<0.01 vs. BDL; $$P<0.01,
$P<0.05 vs. HE. HO-1, heme oxygenase-1; BDL, bile
duct ligation; HE, hepatic encephalopathy; CoPP, cobalt
protoporphyrin; ZnPP, zinc protoporphyrin; COHb,
carboxyhemoglobin. |
Decreased levels of AST, ALT and TBIL were observed
in the ZnPP group compared with the HE group; however, these were
elevated in the CoPP group (Fig.
1B). The levels of ammonia were lower in the ZnPP treatment
group compared with the HE group; however, were higher in the CoPP
treatment group (P<0.01; Fig.
1Cf). The levels of ammonia in the brain were significantly
higher in the HE group compared with the BDL treatment group;
however, these were significantly elevated and reduced in the CoPP
and ZnPP treatment groups, respectively, compared with the HE group
(P<0.01; Fig. 2Cc).
Liver and brain H&E staining demonstrated less
severe fibrous hyperplasia and cerebral intracellular edema in the
brain following treatment with ZnPP; however, these were more
severe in the CoPP group (Fig.
1A). The histopathological scores for liver fibrosis were
higher in HE and CoPP groups than for those of the ZnPP treatment
group (Fig. 1Ad).
Locomotor activity in the BDL group was reduced
compared with the sham controls. In addition, the locomotor
activity was significantly decreased in the HE group compared with
the BDL and CoPP groups (P<0.01); however, this was normalized
following ZnPP treatment (Fig.
3Bb).
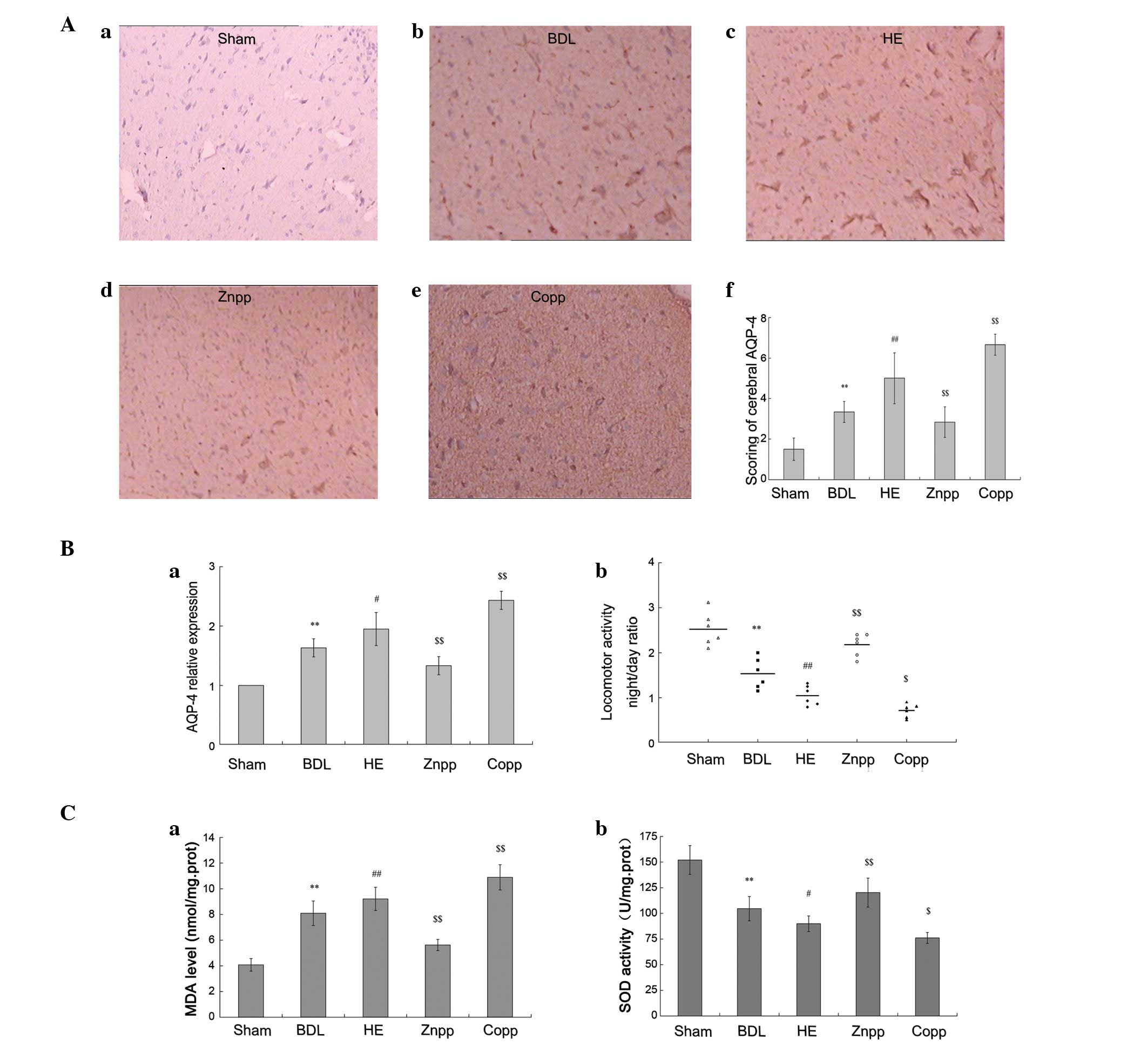 | Figure 3Levels of AQP-4 expression and
oxidative stress in the brain. (A) Immunohistochemical staining
with AQP-4. Brain sections were stained with AQP-4 antibody; (Aa)
AQP-4 was expressed at low levels in the sham group; (Ab, Ac, Ad
and Ae) more intense immunolabeling of the brain cerebral cortex
was observed in the HE and CoPP groups compared with the BDL group;
however, was decreased in the ZnPP group compared with the HE
group; (Af) quantitative scoring of immunohistochemical staining of
cerebral AQP-4 protein expression (original magnification, ×400).
(B) AQP-4 mRNA levels and locomotor activity; (Ba) AQP-4 mRNA
levels were quantified using real-time PCR in brain tissue. The
levels were significantly increased in the HE and CoPP groups
compared with the BDL group, and decreased in the ZnPP group; (Bb)
locomotor activity was recorded prior to the rats being sacrificed.
Data are expressed as the night/day ratio of cumulative distance
traveled, recorded over a 12 h active period (night) and a 12 h
inactive period (day). (C) MDA and SOD levels. Higher MDA and lower
SOD levels were detected in the HE and CoPP groups compared with
the BDL group. However, the (Ca) MDA levels decreased and (Cb) SOD
levels increased significantly in the ZnPP treatment group. The
data are presented as the mean ± SD. **P<0.01 vs.
sham; ##P<0.01, #P<0.05 vs. BDL;
$$P<0.01, $P<0.05 vs. HE. AQP-4,
aquaporin-4; MDA, malonaldehyde; SOD, superoxide dismutase; HE,
hepatic encephalopathy; CoPP, cobalt protoporphyrin; ZnPP, zinc
protoporphyrin; BDL, bile duct ligation. |
The inhibition of HO-1 expression led to decreased
levels of AQP-4 expression and brain edema. The mRNA levels of
AQP-4 in the brain were higher in the BDL group compared with the
sham group and elevated in the HE group compared with the BDL group
(P<0.01). Compared with the HE group, AQP-4 expression was
decreased in the ZnPP treatment group; however, this was
significantly increased in the CoPP treatment group (P<0.01;
Fig. 3Ba).
Cerebral AQP-4 immunostaining demonstrated a
markedly more intense immunolabeling of the cerebral cortex in the
BDL, HE and CoPP treatment groups compared with the sham controls
(Fig. 3Aa-e). Quantitative scoring
of cerebral AQP-4 protein expression demonstrated that it was
reduced in the ZnPP treatment group compared rats in the HE group
(Fig. 3Af).
The water content in the brain significantly
increased in the HE group compared with the BDL treatment group. It
decreased markedly in the ZnPP treatment group compared with the HE
group (P<0.01) and was elevated in the CoPP treatment group
(Fig. 2Cb).
Role of oxidative stress in HE
The levels of oxidative stress in the brain were
assessed by measuring the levels of MDA and SOD. The level of MDA
was evidently higher in the HE group compared with the BDL group;
however, this was lower in the ZnPP treatment group and higher in
the CoPP treatment group compared with the HE group (P<0.01;
Fig. 3Ca). The levels of SOD were
elevated in the BDL group compared with the sham control
(P<0.01) and were significantly increased in the ZnPP treatment
group compared with the HE group (P<0.01); however, these were
decreased in the CoPP treatment group (Fig. 3Cb).
Discussion
HE is an extremely common neuropsychiatric disorder
observed in patients with advanced liver disease. Current treatment
strategies of HE mainly aim to reduce the production and intestinal
absorption of ammonia (2). In the
present study, we demonstrated that treatment with ZnPP improves HE
and alleviates liver fibrosis induced by a hyperammonemic diet in
BDL rats. At 4 weeks, brain edema, hyperammonemia and reduced
locomotor activity were observed in the HE group. Compared with the
HE group, these indicators were ameliorated in the ZnPP group;
however, were more severe in the CoPP group. These results
demonstrated that HO-1 may be important in the induction of HE and
its inhibition may provide an alternative treatment strategy for
HE.
The HO/CO pathway is important in the modulation of
microcirculation in normal and cirrhotic conditions (19). However, the mechanism by which it
affects the pathogenesis of HE remains unknown. In normal
conditions in the central nervous system, the level of HO-1
expression is low (20). Several
studies have demonstrated that HO-1 mRNA expression levels are
elevated in the frontal cortex of HE rats, which exerts a number of
important effects on the coma stage of encephalopathy induced by
liver injury (14,21,22).
In vitro, HO-1 has been shown to stimulate cytoprotection
against oxidative stress (23);
however, this protective action has yet to be uniformly observed
in vivo. Wang and Doré (24) reported that HO-1 is capable of
exacerbating early brain injury following intracerebral hemorrhage.
In addition, non-specific HO inhibitors were able to attenuate
brain injury (25). At a specific
threshold, HO-1 expression may be beneficial and aid in the
protection of hepatocytes (26),
whereas HO-1 overexpression may stimulate or aggravate liver
cirrhosis (27). The
overexpression of HO-1 in the brain cortex of rats acutely
intoxicated with ammonia may contribute to cerebral hyperemia in
hyperammonemic states (28). The
inhibition of bilirubin formation by targeting HO-1 through small
interfering RNA (siRNA) may prevent and treat bilirubin
encephalopathy at an early clinical stage (29). In the present study, the inhibition
of HO-1 was capable of attenuating the levels of serum ammonia and
brain edema. Thus, the inhibition of HO-1 may provide an
alternative strategy to prevent and treat HE.
The predominant endogenous source of CO in the body
derives from the degradation of heme catalyzed by HO-1, forming CO,
biliverdin and free iron molecules (30,31).
COHb levels may be used to estimate the activity of HO in
experimental animals (32). Our
previous studies have demonstrated that the level of COHb is higher
in cirrhotic patients compared with healthy individuals and
patients with chronic hepatitis. These findings have additionally
been reported in several other studies (33–35).
The overexpression of CO leads to the over-expansion of splanchnic
vessels, a decrease in vascular resistance and an increase in blood
volume, leading to the hyperdynamic circulation of hepatic
cirrhosis with portal hypertension (36,37).
In the present study, serum COHb levels increased in the HE and
CoPP treatment groups with portal hypertension and brain injury.
However, HE was alleviated in the ZnPP treatment group, which
demonstrated lower levels of serum COHb. This may be due to the
inhibition of HO-1 expression decreasing PVP by regulating the
CO/NO pathway and reducing the levels of toxic substances
transported into the brain. The HO/CO pathway may be involved in
the pathogenesis of HE in addition to a number of other known
mechanisms. Previous studies have demonstrated that the
simultaneous inhibition of NO and HO completely reverses the
reduction in the vasoconstrictor response to potassium chloride in
the mesenteric vascular bed (36).
We hypothesize that splanchnic vasodilation presenting in portal
hypertension is likely to be multifactorial in origin and
stimulated in part by an excessive release of NO, CO and several
other vasoactive mediators.
Free iron is a byproduct of the degradation of heme
catalyzed by HO-1. Free iron molecules are capable of participating
in the Fenton and Haber-Weiss reactions, and excessive redox-active
iron may lead to oxidative stress and the subsequent damage of
membranes, proteins and DNA (38).
Notably, the overexpression of HO-1 did not lead to an overload of
iron in the brain cortex and other anatomical sites, indicated in
our study by the Perl’s Prussian blue stain (data not shown). This
was due to iron primarily accumulating in reticuloendothelial
cells, including the spleen, liver and bone. In addition, the
overproduction of iron may transfer to other organs apart for the
brain, avoiding injury to cerebral cells.
Brain edema is an important component of HE that is
associated with acute liver failure. The edema is mainly due to the
swelling of astrocytes (cytotoxic edema). Ammonia and AQP-4 are
abundantly expressed in astrocytes and may be involved in the
development of edema (39). AQP-4
is the predominant water channel expressed in the brain and is
distributed widely in brain tissue, with the exception of neurons.
It has been demonstrated that AQP-4 is important in maintaining
water homeostasis in the brain tissue and participates in
super-acute ischemic brain edema (40,41).
Early cytotoxic brain edema following brain injury in addition to a
secondary insult or focal ischemia results in a vasopressin V
receptor-mediated response, most likely through the upregulation of
AQP-4 (42). In the present study,
the expression levels of AQP-4 and HO-1 were increased in the HE
and BDL groups. The overexpression of HO-1 in the CoPP treatment
group led to a high brain water content, which may be correlated
with the activation of AQP-4 by ammonia. By contrast, lower levels
of ammonia in plasma due to low PVP in the ZnPP treatment group led
to the inhibition of AQP-4 expression and a reduction in the
severity of brain edema.
Several studies have demonstrated an increased level
of oxidative/nitrosative stress in the brain of liver failure
patients and its reduction has been shown to be beneficial in
animal models (43).
Oxidative/nitrosative stress is important in ammonia-induced cell
swelling in cultured cells and SOD inhibits the swelling of
astrocytes. Wang et al(24)
demonstrated that HO-1 knockout mice sustain less severe brain
injury and exhibit less neurological dysfunction compared with
wild-type mice during the early stages following intracerebral
hemorrhage. Previous studies have demonstrated that excess iron is
able to participate in the Fenton and Haber-Weiss reactions,
leading to oxidative stress and subsequent damage to cell
membranes, proteins and DNA (38).
In the present study, the upregulation of HO-1 expression increased
the levels of oxidative stress, which may be due to free iron
produced from the degradation of heme catalyzed by HO-1. Inhibition
of HO-1 in the ZnPP treatment group led to lower levels of
oxidative stress in the brain and a notable reduction in the levels
of neurocyte injury.
In conclusion, we demonstrated that the inhibition
of HO-1 expression is capable of attenuating HE by inhibiting the
expression of APQ-4 and reducing the levels of oxidative stress,
while decreasing PVP and improving liver fibrosis in BDL rats. The
results additionally demonstrated that the HO/CO pathway is
important in the inhibition of HO-1 expression, which may prevent
HE, and elucidate the role of high levels of COHb in cirrhosis
patients with HE (11). Brain
biopsies are rarely performed in patients with HE due to the
temporary nature of the disease and surgery often not being
necessary; therefore, the animal experiment provides an appropriate
supplement to the cerebral pathology of patients with HE. In
addition, we demonstrated that HO-1 is important in a number of
organs involved in end-stage liver disease (9,10).
Thus, additional research with selective HO inhibitors and their
doses is required. Effectively regulating the levels of HO-1 during
liver cirrhosis may be beneficial. In addition, we hypothesize that
a reduction in the levels of CO resulting from the inhibition of
HO-1 may provide a novel therapeutic strategy for decreasing PVP
and reducing plasma ammonia levels. The HO/CO pathway is involved
in the regulation of HE pathogenesis and may therefore provide a
novel therapy for HE.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 30970886), the Science,
Technology Project of Dalian (no. 2010E15SF179) and the Doctoral
Initial Funding of Liaoning province (no. 20121110).
References
|
1
|
Ferenci P, Lockwood A, Mullen K, Tarter R,
Weissenborn K and Blei AT: Hepatic encephalopathy - definition,
nomenclature, diagnosis, and quantification: final report of the
working party at the 11th World Congresses of Gastroenterology,
Vienna, 1998. Hepatology. 35:716–721. 2002. View Article : Google Scholar
|
|
2
|
Atluri DK, Prakash R and Mullen KD:
Pathogenesis, diagnosis, and treatment of hepatic encephalopathy. J
Clin Exp Hepatol. 2:77–86. 2011. View Article : Google Scholar
|
|
3
|
Sass G, Barikbin R and Tiegs G: The
multiple functions of heme oxygenase-1 in the liver. Z
Gastroenterol. 50:34–40. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barañano DE and Snyder SH: Neural roles
for heme oxygenase: contrasts to nitric oxide synthase. Proc Natl
Acad Sci USA. 98:10996–11002. 2001.PubMed/NCBI
|
|
5
|
Turner CP, Bergeron M, Matz P, et al: Heme
oxygenase-1 is induced in glia throughout brain by subarachnoid
hemoglobin. J Cereb Blood Flow Metab. 18:257–273. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matz PG, Weinstein PR and Sharp FR: Heme
oxygenase-1 and heat shock protein 70 induction in glia and neurons
throughout rat brain after experimental intracerebral hemorrhage.
Neurosurgery. 40:152–160. 1997.PubMed/NCBI
|
|
7
|
Chao XD, Ma YH, Luo P, et al:
Up-regulation of heme oxygenase-1 attenuates brain damage after
cerebral ischemia via simultaneous inhibition of superoxide
production and preservation of NO bioavailability. Exp Neurol.
239:163–169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cuadrado A and Rojo AI: Heme oxygenase-1
as a therapeutic target in neurodegenerative diseases and brain
infections. Curr Pharm Des. 14:429–442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo SB, Duan ZJ, Li Q and Sun XY: Effect
of heme oxygenase-1 on renal function in rats with liver cirrhosis.
Shi Jie Wei Chang Bing Xue Za Zhi Bian Ji Bu. 17:322–328. 2011.(In
Chinese).
|
|
10
|
Guo SB, Duan ZJ, Li Q and Sun XY: Effects
of heme oxygenase-1 on pulmonary function and structure in rats
with liver cirrhosis. Zhong Hua Yi Xue Za Zhi Bian Ji Bu.
124:918–922. 2011.(In Chinese).
|
|
11
|
Sun XY, Duan ZJ, Li YL and Chang QS:
Detection of carboxyhemoglobin in patients with hepatic
encephalopathy due to hepatitis B virus-related cirrhosis. Zhong
Hua Yi Xue Za Zhi Bian Ji Bu. 125:3991–3996. 2012.(In Chinese).
|
|
12
|
Jover R, Rodrigo R, Felipo V, et al: Brain
edema and inflammatory activation in bile duct ligated rats with
diet-induced hyperammonemia: A model of hepatic encephalopathy in
cirrhosis. Hepatology. 43:1257–1266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Amersi F, Buelow R, Kato H, Ke B, Coito
AJ, Shen XD, Zhao D, Zaky J, Melinek J, Lassman CR, Kolls JK, Alam
J, Ritter T, Volk HD, Farmer DG, Ghobrial RM, Busuttil RW and
Kupiec-Weglinski JW: Upregulation of heme oxygenase-1 protects
genetically fat Zucker rat livers from ischemia/reperfusion injury.
J Clin Invest. 104:1631–1639. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang W, Desjardins P and Butterworth RF:
Hypothermia attenuates oxidative/nitrosative stress, encephalopathy
and brain edema in acute (ischemic) liver failure. Neurochem Int.
55:124–128. 2009. View Article : Google Scholar
|
|
15
|
Butterworth RF, Lalonde R, Power C, Baker
GB, Gamrani H and Ahboucha S: Dehydroepiandrosterone sulphate
improves cholestasis-associated fatigue in bile duct ligated rats.
Neurogastroenterol Motil. 21:1319–1325. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shackelford C, Long G, Wolf J, Okerberg C
and Herbert R: Qualitative and quantitative analysis of
nonneoplastic lesions in toxicology studies. Toxicol Pathol.
30:93–96. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Remmele W and Stegner HE: Recommendation
for uniform definition of an immunoreactive score (IRS) for
immunohistochemical estrogen receptor detection (ER-ICA) in breast
cancer tissue. Pathologe. 8:138–140. 1987.(In German).
|
|
18
|
Kakinuma S, Tanaka Y, Chinzei R, et al:
Human umbilical cord blood as a source of transplantable hepatic
progenitor cells. Stem Cells. 21:217–227. 2003.PubMed/NCBI
|
|
19
|
Van Landeghem L, Laleman W, Vander Elst I,
et al: Carbon monoxide produced by intrasinusoidally located
haem-oxygenase-1 regulates the vascular tone in cirrhotic rat
liver. Liver Int. 29:650–660. 2009.PubMed/NCBI
|
|
20
|
Chopra VS, Chalifour LE and Schipper HM:
Differential effects of cysteamine on heat shock protein induction
and cytoplasmic granulation in astrocytes and glioma cells. Brain
Res Mol Brain Res. 31:173–184. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jiang W, Desjardins P and Butterworth RF:
Minocycline attenuates oxidative/nitrosative stress and cerebral
complications of acute liver failure in rats. Neurochem Int.
55:601–605. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chastre A, Jiang W, Desjardins P and
Butterworth RF: Ammonia and proinflammatory cytokines modify
expression of genes coding for astrocytic proteins implicated in
brain edema in acute liver failure. Metab Brain Dis. 25:17–21.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen K, Gunter K and Maines MD: Neurons
overexpressing heme oxygenase-1 resist oxidative stress-mediated
cell death. J Neurochem. 75:304–313. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang J and Doré S: Heme oxygenase-1
exacerbates early brain injury after intracerebral haemorrhage.
Brain. 130:1643–1652. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koeppen AH, Dickson AC and Smith J: Heme
oxygenase in experimental intracerebral hemorrhage: the benefit of
tin-mesoporphyrin. J Neuropathol Exp Neurol. 63:587–597.
2004.PubMed/NCBI
|
|
26
|
Malaguarnera L, Madeddu R, Palio E, Arena
N and Malaguarnera M: Heme oxygenase-1 levels and oxidative
stress-related parameters in non-alcoholic fatty liver disease
patients. J Hepatol. 42:585–591. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Froh M, Conzelmann L, Walbrun P, et al:
Heme oxygenase-1 overexpression increases liver injury after bile
duct ligation in rats. Shi Jie Wei Chang Bing Xue Za Zhi Bian Ji
Bu. 13:3478–3486. 2007.(In Chinese).
|
|
28
|
Warskulat U, Görg B, Bidmon HJ, Müller HW,
Schliess F and Häussinger D: Ammonia-induced heme oxygenase-1
expression in cultured rat astrocytes and rat brain in vivo. Glia.
40:324–336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xia ZW, Li CE, Jin YX, et al: Reduction of
bilirubin by targeting human heme oxygenase-1 through siRNA. Exp
Biol Med (Maywood). 232:495–502. 2007.PubMed/NCBI
|
|
30
|
De las Heras D, Fernändez J, Ginès P, et
al: Increased carbon monoxide production in patients with cirrhosis
with and without spontaneous bacterial peritonitis. Hepatology.
38:452–459. 2003.PubMed/NCBI
|
|
31
|
Tran TT, Martin P, Ly H, Balfe D and
Mosenifar Z: Carboxyhemoglobin and its correlation to disease
severity in cirrhotics. J Clin Gastroenterol. 41:211–215. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carter EP, Hartsfield CL, Miyazono M,
Jakkula M, Morris KG Jr and McMurtry IF: Regulation of heme
oxygenase-1 by nitric oxide during hepatopulmonary syndrome. Am J
Physiol Lung Cell Mol Physiol. 283:L346–L353. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu J, Duan J, Zhu L, Yang D, Wang L and
Shao H: The preliminary study of the relationship between
oxygenase-carbon monoxide system and plasma endothelin in hepatic
cirrhotic patients with portal hypertension. Zheng Zhou Da Xue.
9:745–748. 2008.(In Chinese).
|
|
34
|
Tarquini R, Masini E, La Villa G, et al:
Increased plasma carbon monoxide in patients with viral cirrhosis
and hyperdynamic circulation. Am J Gastroenterol. 104:891–897.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen YC, Ginès P, Yang J, et al: Increased
vascular heme oxygenase-1 expression contributes to arterial
vasodilation in experimental cirrhosis in rats. Hepatology.
39:1075–1087. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fernandez M, Lambrecht RW and Bonkovsky
HL: Increased heme oxygenase activity in splanchnic organs from
portal hypertensive rats: role in modulating mesenteric vascular
reactivity. J Hepatol. 34:812–817. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Angermayr B, Mejias M, Gracia-Sancho J,
Garcia-Pagan JC, Bosch J and Fernandez M: Heme oxygenase attenuates
oxidative stress and inflammation, and increases VEGF expression in
portal hypertensive rats. J Hepatol. 44:1033–1039. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pietrangelo A: Metals, oxidative stress,
and hepatic fibrogenesis. Semin Liver Dis. 16:13–30. 1996.
View Article : Google Scholar
|
|
39
|
Rama Rao KV and Norenberg MD: Aquaporin-4
in hepatic encephalopathy. Metab Brain Dis. 22:265–275.
2007.PubMed/NCBI
|
|
40
|
Papadopoulos MC and Verkman AS:
Aquaporin-4 and brain edema. Pediatr Nephrol. 22:778–784. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Badaut J, Ashwal S, Adami A, et al: Brain
water mobility decreases after astrocytic aquaporin-4 inhibition
using RNA interference. J Cereb Blood Flow Metab. 31:819–831. 2011.
View Article : Google Scholar
|
|
42
|
Kleindienst A, Dunbar JG, Glisson R and
Marmarou A: The role of vasopressin V1A receptors in cytotoxic
brain edema formation following brain injury. Acta Neurochir
(Wien). 155:151–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bemeur C, Desjardins P and Butterworth RF:
Evidence for oxidative/nitrosative stress in the pathogenesis of
hepatic encephalopathy. Metab Brain Dis. 25:3–9. 2010. View Article : Google Scholar : PubMed/NCBI
|














