Introduction
Mitochondrial DNA depletion syndrome (MTDPS)
consists of a group of genetically and clinically heterogeneous
disorders caused by nuclear-mitochondrial intergenomic defects
characterized by a significant reduction in mtDNA content (1–4).
MTDPS is divided into several types (from MTDPS1 to MTDPS10)
according to the distinct clinical phenotypes and genetic causes.
The clinical phenotypes frequently overlap with each other, and
thus require an exact diagnosis in order to distinguish each type.
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), a
frequently diagnosed type of MTDPS, is an autosomal recessive
multiorgan disorder characterized by gastrointestinal (GI)
dysmotility, cachexia, myopathy, peripheral neuropathy,
leukoencephalopathy and mitochondrial dysfunction. To date, three
subtypes of MNGIE have been described; MTDPS1 (MIM# 603041) caused
by the thymidine phosphorylase (TYMP) gene on
22q13.33 (5), MTDPS4B (MIM#
613662) caused by DNA polymerase γ (POLG) on 15q26.1
(6,7) and MTDPS8B (MIM# 612075) caused by
p53-inducible ribonucleotide reductase small subunit
(RRM2B) on 8q22.3 (8,9).
Mutations in POLG and RRM2B also cause MTDPS4A
(Alpers’ type) and autosomal-dominant progressive external
ophthalmoplegia, respectively (10,11).
Several homozygous or compound heterozygous
mutations in TYMP have been reported to be the underlying
causes of MNGIE (5,10–17).
Thymidine phosphorylase (TP) encoded by TYMP catalyzes the
phosphorylation of thymidine or deoxyuridine to thymine or uracil
in mtDNA synthesis (4). The
protein was originally identified as a platelet-derived endothelial
cell growth factor (PDECGF), an angiogenic factor distinct from the
previously described endothelial cell mitogens of the fibroblast
growth factor family (18). A TP
deficiency is a severe clinical condition in the affected tissues
and is ultimately fatal (19). In
a previous study, the inhibition of TP activity led to elevated
pyrimidine levels and consequential axonal swelling (20). The inhibition of TP activity in
TYMP knockout mice has been shown to result in the depletion
or deletion of mtDNA (4,21). Therefore, mutations in TYMP
are relevant to the impaired replication and maintenance of mtDNA.
On average, the onset of MNGIE in patients with a homozygous
TYMP mutation occurs in the late teens, with death occurring
in the thirties. However, patients with compound heterozygous
TYMP mutations showed incomplete MNGIE phenotypes with a
late onset occurring in the forties or fifties (15–17).
We identified a female Korean MNGIE patient with an
early-onset but mild phenotype. The patient revealed typical MNGIE
phenotypes, including GI discomfort, external ophthalmoplegia,
pigmentary retinopathy and sensory motor polyneuropathy. Molecular
genetic analysis revealed novel compound heterozygous mutations in
TYMP, indicating that these particular mutations may be the
genetic causes of the MNGIE phenotype.
Materials and methods
Subjects
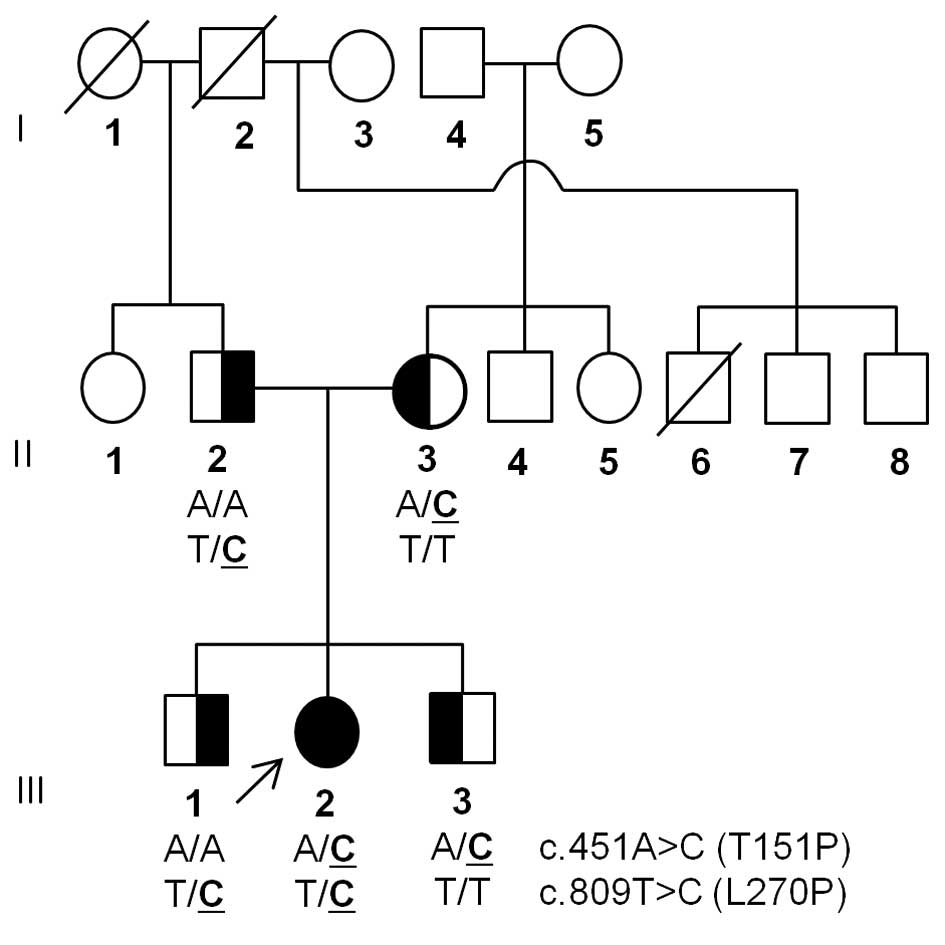
A Korean MNGIE family (family ID: MT119) was
enrolled in this study (Fig. 1).
Furthermore, 225 healthy controls were recruited from the
Neurological Department, Ewha Womans University, Mokdong Hospital
(Seoul, Korea). The paternity of the patient was confirmed by
genotyping 15 microsatellite markers using a PowerPlex 16 kit
(Promega, Madison, WI, USA). This study was approved by the
Institutional Review Board for Ewha Womans University, Mokdong
Hospital and all participants provided written informed
consent.
Clinical and electrophysiological
assessments
Clinical information was obtained by neurological
examination, including the assessment of muscle weakness, sensory
impairment and reflexes. Muscle strength was assessed manually
using the standard Medical Research Council (MRC) scale (http://www.mrc.ac.uk). Nerve conduction studies of the
median, ulnar, fibular, tibial, sural and superficial fibular
nerves were determined. Electromyography (EMG) was performed in the
first dorsal interosseous, biceps brachii, tibialis anterior,
medial gastrocnemius and vastus lateralis muscles. Ultrasound
examination for the peripheral nerves was performed on the
bilateral median, ulnar, radial, sciatic, fibular, tibial and sural
nerves. Blood TP activity was determined spectrophotometrically by
measuring the conversion of thymidine to thymine in an end-point
assay (Laboratory of Personalized Genomic Medicine, Columbia
University, New York, NY, USA).
MRI studies
Whole brain MRIs were obtained from 5-mm slices
without interslice gaps to produce 30 axial images using a 3.0-T
system (Achieva, Philips, Netherlands). The imaging protocol
consisted of T2-weighted spin echo (TR/TE=2,500/80 msec),
T1-weighted spin echo (TR/TE=400/10 msec) and fluid-attenuated
inversion recovery (FLAIR; TR/TE=6,000/120 msec; inversion time,
2,000 msec) images.
Molecular study
Total DNA was extracted from leukocytes using a
QIAamp Blood DNA kit (Qiagen, Hilden, Germany). Entire mtDNA was
amplified by PCR using 46 primer sets from the MitoSEQr
resequencing system (Applied Biosystems, Foster City, CA, USA). All
coding exons of the PEO1, TYMP, ANT1,
POLG1, POLG2, DGUOK, RRM2B and
TK2 genes were amplified using PCR. The primer sequences and
PCR conditions used are available on request. PCR products were
sequenced by an automatic genetic analyzer ABI 3130xl using a
BigDye terminator cycle sequencing ready reaction kit (Applied
Biosystems). Sequence variations were identified by the SeqScape
(ver. 2.1, Applied Biosystems) and Chromas software (ver. 2.33,
Technelysium, South Brisbane, Australia). Sequence variations were
confirmed by analyzing both strands of DNA. cDNA numbering was
achieved with +1 corresponding to the A of the ATG initiation
codon, according to the mutation nomenclature instruction of the
Human Genome Variation Society (http://www.hgvs.org/mutnomen/). In silico
predictions were performed using the SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)
and MUpro (SVM; http://mupro.proteomics.ics.uci.edu/) programs.
Multiple alignments of amino acid sequences among different species
were performed using the MEGA5 program (ver 5.05) (22). Deletion of 4977 bp
(m.8470_13446del4977) in mtDNA, frequently called a ‘common
deletion’, was detected using an Expand Long Template PCR System
(Roche, Mannheim, Germany). The used PCR primers covered the
following regions of the revised Cambridge reference sequence:
8225–8247 (forward) and 13707–13729 (reverse).
Results
Identification of compound heterozygous
mutations in TYMP
Mutation screening revealed two TYMP
heterozygous missense mutations; Thr151Pro (c.451A>C) and
Leu270Pro (c.809T>C; Fig. 2A).
One of these mutations was transmitted from each parent of the
patient; Leu270Pro from the father and Thr151Pro from the mother.
The patient had two unaffected siblings, each receiving a single
mutation from their parents. These mutations were not reported in
the dbSNP137 (http://ncbi.nlm.nih.gov/) and 1000 Genome Database
(http://www.1000genomes.org/), and were
not found in the 225 controls. Both mutation sites were highly
conserved among different species (Fig. 2B). Several in silico
analyses predicted that the mutations affect protein function
(Table I). No other causative
mutation was observed in the examined nuclear genes.
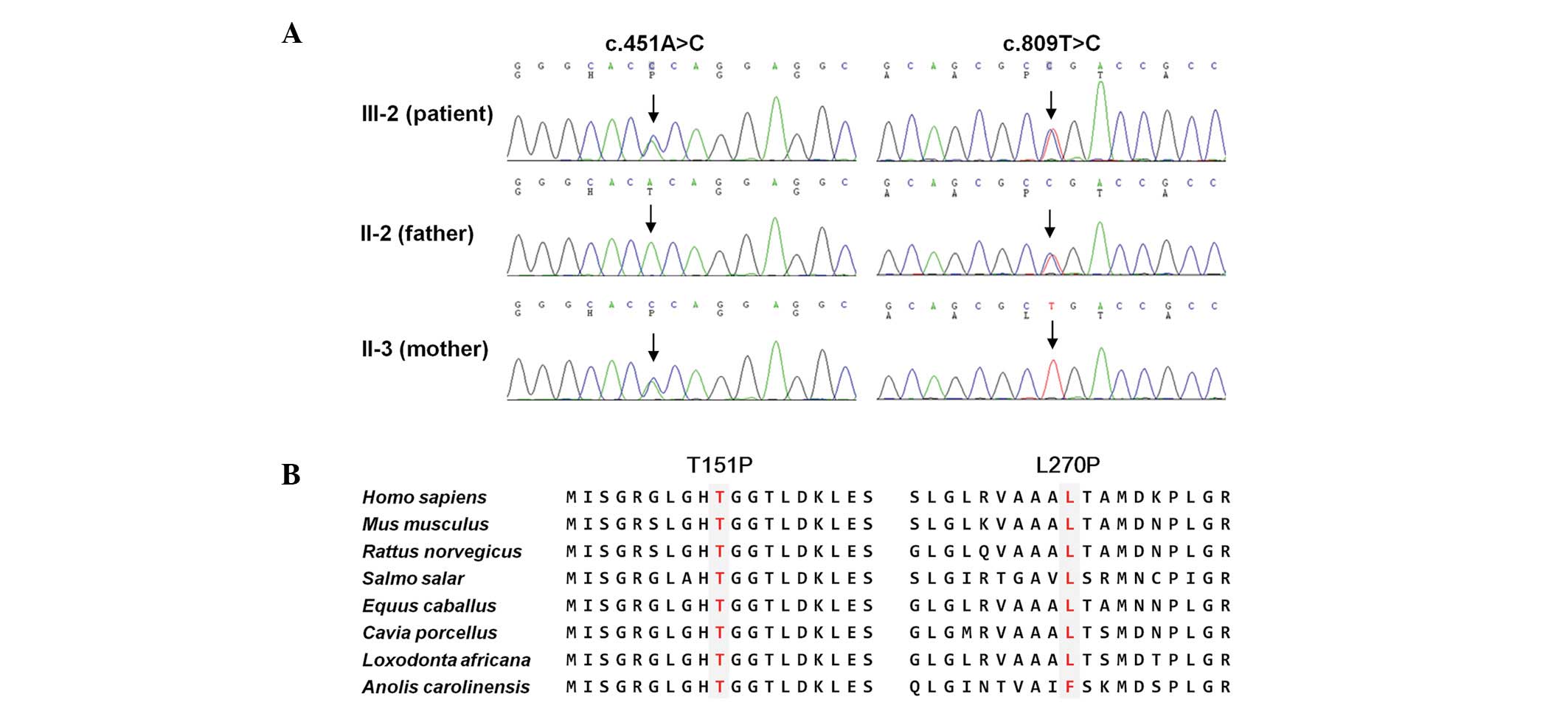 | Figure 2Identification of TYMP compound
heterozygous mutations in a MNGIE family. (A) Sequencing
chromatograms of two mutations. Vertical arrows indicate the
mutation site. The patient (III-2) was found to have both mutations
and the parents of the individual had one of each mutation. (B)
Conservation analysis of amino acid sequences. The analysis was
conducted using the MEGA5 ver 5.05 program. Both mutation sites
were highly conserved between different species (H. sapiens,
NP_001944.1; M. musculus, NP_612175.1; R. norvegicus,
NP_001012122.1; S. salar, NP_001133751.1; E.
caballus, XP_001914955.1; C. porcellus, XP_003461632.1;
L. africana, XP_003423234.1; A. carolinensis,
XP_003228670.1). TYMP, thymidine phosphorylase gene;
MNGIE, mitochondrial nerurogastrointestinal encephalomyopathy. |
 | Table ICompound heterozygous mutations of
TYMP in a MNGIE family. |
Table I
Compound heterozygous mutations of
TYMP in a MNGIE family.
| Family | Substitution | Amino acid | Phenotype | Inheritance | In silico
analysis (S/P2/M)b |
|---|
|
|---|
| Gene | Nucleotidea |
|---|
| MT119 | TYMP | c.451A>C | T151P | MNGIE | Recessive | 0.00c/1.00c/1 |
| | c.809T>C | L270P | | | 0.00c/1.00c/−1c |
Identification of mtSNPs
Whole mtDNA sequencing revealed numerous
mitochondrial single nucleotide polymorphisms (mtSNPs; Table II). However, all the mtSNPs have
been reported to be polymorphic in the MITOMAP-Human Mitochondrial
Genome Database (http://mitomap.org/MITOMAP) or the mtDB-Human
Mitochondrial Genome Database (http://www.mtdb.igp.uu.se) (24). Although ATP6 m.8794C>T
(His90Tyr) has been associated with high-performance endurance
running (25), it was not
considered to be causative for MNGIE, as it was observed in the
controls and has also been reported to be a polymorphic mtSNP in
the MITOMAP. The long template PCR from the blood DNA of the
patient revealed no common large deletion of mtDNA
(m.8470_13446del4977).
| Table IIVariants identified from whole mtDNA
in the MNGIE patient (III-2). |
Table II
Variants identified from whole mtDNA
in the MNGIE patient (III-2).
| Gene | Nt positiona | Nucleotide
change | Amino acid
change |
Characterization | Report |
|---|
| D-loop | 73 | A>G | - | Poly | Yes |
| 152 | T>C | - | Poly | Yes |
| 200 | A>G | - | Poly | Yes |
| 263 | A>G | - | Poly | Yes |
| 315.1 | C insertion | - | Poly | Yes |
| 523 | A deletion | - | Poly | Yesb |
| 524 | C deletion | - | Poly | Yesb |
| 12S
rRNA | 663 | A>G | - | Poly | Yes |
| 750 | A>G | - | Poly | Yes |
| 1438 | A>G | - | Poly | Yes |
| 16S
rRNA | 1736 | A>G | - | Poly | Yes |
| 2706 | A>G | - | Poly | Yes |
| ND1 | 4248 | T>C | Syn | Poly | Yes |
| ND2 | 4769 | A>G | Syn | Poly | Yes |
| 4824 | A>G | Thr119Ala | Poly | Yes |
| CO1 | 6060 | A>G | Ile53Val | Poly | Yes |
| 7028 | C>T | Syn | Poly | Yes |
| ATP6 | 8794 | C>T | His90Tyr | Poly/exercise
endurance | Yes |
| 8860 | A>G | Thr112Ala | Poly | Yes |
| ND4 | 11719 | G>A | Syn | Poly | Yes |
| ND5 | 12705 | C>T | Syn | Poly | Yes |
| CytB | 14766 | C>T | Thr7Ile | Poly | Yes |
| 15326 | A>G | Thr194Ala | Poly | Yes |
| D-loop | 16223 | C>T | - | Poly | Yes |
| 16290 | C>T | - | Poly | Yes |
| 16319 | G>A | - | Poly | Yes |
| 16362 | T>C | - | Poly | Yes |
Clinical manifestations
A 28-year-old female patient was referred to Kangbuk
Samsung Hospital (Seoul, Korea) due to abdominal pain, diarrhea,
fever (≤39.5°C) and headaches. Computed tomography (CT) scans
revealed a diffuse white matter change in the brain. In early
childhood, the patient frequently suffered from abdominal pain,
diarrhea and vomiting approximately once a week, which was
aggravated by febrile illness. Since elementary school, the
patient’s capacity for exercise has been decreased. In high school,
the patient had cosmetic surgery to correct bilateral ptosis. At 21
years old the patient had an eye operation to correct strabismus,
but did not suffer from diplopia. One year ago, the patient was
diagnosed with fatty liver and polycystic ovary syndrome. Upon
admission, the patient’s vital signs were stable with a BMI of 18
kg/m2 (height 165 cm and weight 49 kg). The patient did
not appear to exhibit cachexia.
Neurological examination revealed mild bilateral
ptosis, mild bilateral external ophthalmoparesis and areflexia of
stretch reflexes; however, there was no cognitive dysfunction or
motor or sensory abnormalities. Pigmentary retinopathy was observed
in both eyes upon fundus examination. Nerve conduction study
revealed demyelinating type diffuse sensory motor polyneuropathy
and there were conduction blocks on bilateral tibial nerves. Needle
EMG revealed normal insertional and spontaneous activity, and
neurogenic motor unit potential without myogenic potential.
Ultrasonographic study of the peripheral nerves in all four
extremities revealed mild nerve enlargement of bilateral median
nerves. Somatosensory evoked potential studies revealed peripheral
conduction defects. A visual evoked potential study demonstrated
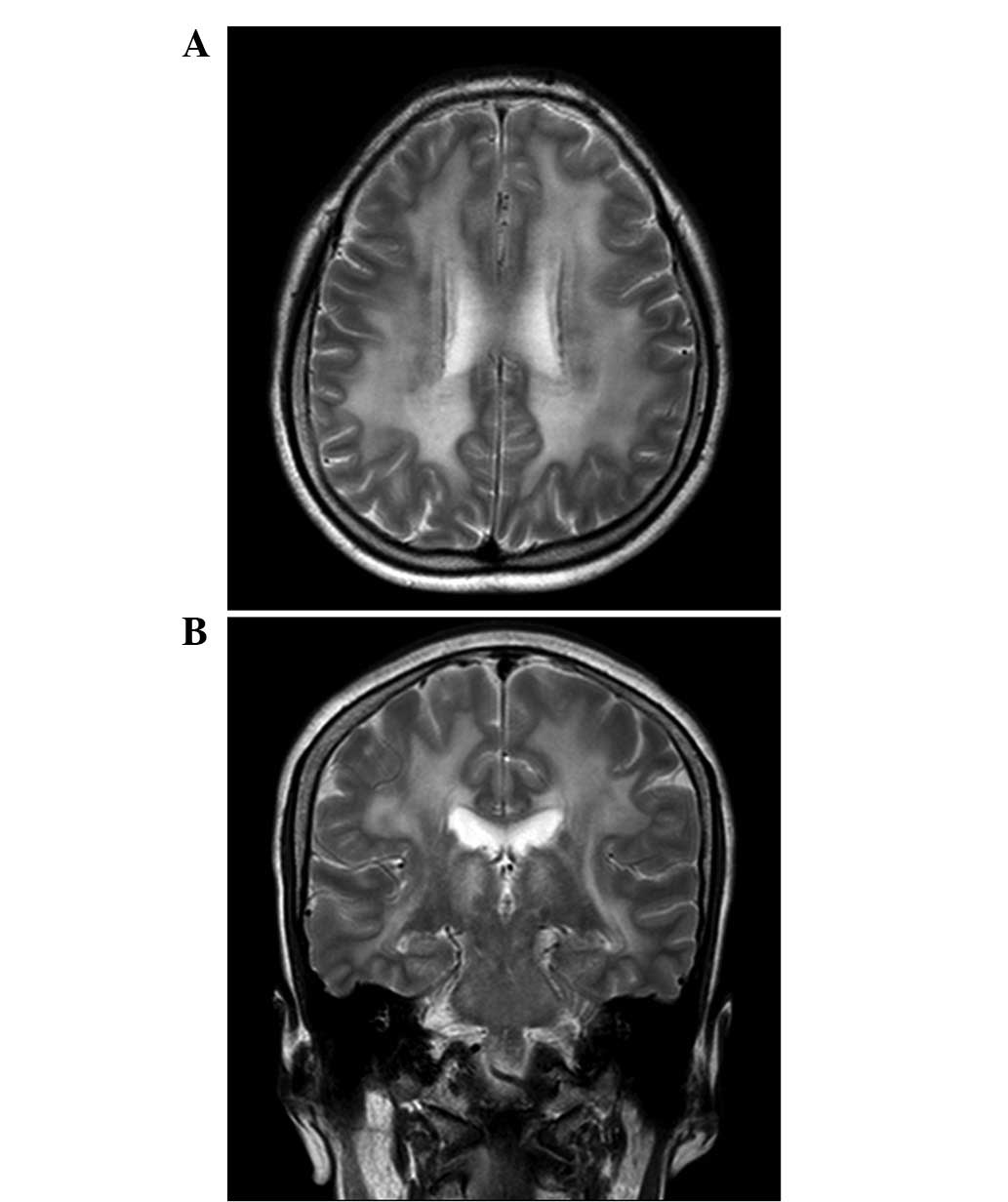
conduction delay. An MRI revealed diffuse white matter high signal
intensity with slight involvement of the deep nuclei, brainstem and
cerebellum; however, MR spectroscopy did not reveal any
abnormalities (Fig. 3).
In the laboratory study, the level of creatine
kinase was 1240 IU/l, serum lactic acid was 1.98 mmol/l (normal,
0.5–2.2) and pyruvic acid was 0.038 mmol/l (normal, 0.034–0.102),
and the cerebrospinal fluid study was normal. Examinations for
leukodystrophy (arylsulfatase A, N-acetyl aspartic acid,
β-galactocerebrosidase and phytanic acid) revealed normal levels.
Buffy coat TP activity decreased to 9.6% of the normal level
(patient, 61 nmol/h/mg protein; normal, 634±217 nmol/h/mg
protein).
Discussion
The present study reports an autosomal recessive
early-onset but mild phenotype MNGIE patient with compound
heterozygous TYMP mutations Thr151Pro (c.451A>C) and
Leu270Pro (c.809T>C). Since i) these mutations were not found in
healthy controls and no unaffected familial member had both
mutations; ii) the mutation sites were well conserved; iii) both
mutations caused an amino acid substitution with Pro residue which
frequently acts as a structural disruptor of protein secondary
structure such as alpha helix; and iv) no other causative mutations
were identified in the nuclear genes or the mtDNA, we hypothesize
that the two identified heterozygous mutations may be the
underlying cause of MNGIE.
The TYMP gene contains 10 exons spanning
>4.3 kb (25) and encodes TP,
which catalyzes the phosphorolysis of thymidine, an essential step
in the nucleotide salvage pathway for mtDNA replication (4). The 482-residue TP has a molecular
mass of 49.97 kD and usually occurs as a homodimeric structure
(26).
To date, several homozygous and compound
heterozygous TYMP mutations have been reported to be
causative of autosomal recessive MNGIE (5,12–17).
Homozygous mutations in TYMP usually result in an almost
complete abolition of TP activity, with <5% remaining (5,13,14);
however, compound heterozygous mutations exhibit a partial loss of
activity with 10–20% normal TP activity (15,16).
Although heterozygous carriers are clinically asymptomatic, TP
activity is decreased to 26–35% of the normal level (15). Patients with compound heterozygous
mutations have been reported to have mild symptoms with a later
onset than that found in typical homozygous patients (15,16).
In an Italian family, compound heterozygous mutations in
TYMP exhibited different clinical presentations between two
affected siblings (17).
With the exception of cachexia, the patient suffered
from the majority of symptoms associated with MNGIE, including
frequent GI discomfort, external ophthalmoplegia, pigmentary
retinopathy and demyelinating type diffuse sensory motor
polyneuropathy. The TP activity of the patient was determined to be
9.6% of the normal value, which corresponds to an intermediate
level between that of the mild phenotype of late-onset heterozygous
patients and that of the severe cachexic phenotype of homozygous
patients (5,13).
To the best of our knowledge, this is the first
report of a Korean MNGIE patient with compound heterozygous
mutations in TYMP. The patient revealed an early-onset but
mild phenotype; therefore, patients with heterozygous mutations may
exhibit mild phenotypes with variable onset ages, partially based
on the reduction in levels of TP activity.
Acknowledgements
This study was supported by a grant from the Korean
Health Technology R&D Project, Ministry of Health & Welfare
(A120814) and the National Research Foundation (NRF) grant, funded
by the Ministry of Education, Science and Technology, Republic of
Korea (2011-0013694).
References
|
1
|
Spelbrink JN, Li FY, Tiranti V, et al:
Human mitochondrial DNA deletions associated with mutations in the
gene encoding Twinkle, a phage T7 gene 4-like protein localized in
mitochondria. Nat Genet. 28:223–231. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirano M, Nishigaki Y and Martí R:
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a
disease of two genomes. Neurologist. 10:8–17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Spinazzola A and Zeviani M: Disorders of
nuclear-mitochondrial intergenomic signaling. Gene. 354:162–168.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suomalainen A and Isohanni P:
Mitochondrial DNA depletion syndromes - many genes, common
mechanisms. Neuromuscul Disord. 20:429–437. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nishino I, Spinazzola A and Hirano M:
Thymidine phosphorylase gene mutations in MNGIE, a human
mitochondrial disorder. Science. 283:689–692. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vissing J, Ravn K, Danielsen ER, et al:
Multiple mtDNA deletions with features of MNGIE. Neurology.
59:926–929. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Van Goethem G, Schwartz M, Löfgren A, et
al: Novel POLG mutations in progressive external ophthalmoplegia
mimicking mitochondrial neurogastrointestinal encephalomyopathy.
Eur J Hum Genet. 11:547–549. 2003.
|
|
8
|
Shaibani A, Shchelochkov OA, Zhang S, et
al: Mitochondrial neurogastrointestinal encephalopathy due to
mutations in RRM2B. Arch Neurol. 66:1028–1032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kollberg G, Darin N, Benan K, et al: A
novel homozygous RRM2B missense mutation in association with severe
mtDNA depletion. Neuromuscul Disord. 19:147–150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Naviaux RK and Nguyen KV: POLG mutations
associated with Alpers’ syndrome and mitochondrial DNA depletion.
Ann Neurol. 55:706–712. 2004.
|
|
11
|
Tyynismaa H, Ylikallio E, Patel M, Molnar
MJ, Haller RG and Suomalainen A: A heterozygous truncating mutation
in RRM2B causes autosomal-dominant progressive external
ophthalmoplegia with multiple mtDNA deletions. Am J Hum Genet.
85:290–295. 2009. View Article : Google Scholar
|
|
12
|
Gamez J, Ferreiro C, Accarino ML, et al:
Phenotypic variability in a Spanish family with MNGIE. Neurology.
59:455–457. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Szigeti K, Wong LJ, Perng CL, et al: MNGIE
with lack of skeletal muscle involvement and a novel TP splice site
mutation. J Med Genet. 41:125–129. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hirano M, Lagier-Tourenne C, Valentino ML,
Martí R and Nishigaki Y: Thymidine phosphorylase mutations cause
instability of mitochondrial DNA. Gene. 354:152–156. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Martí R, Verschuuren JJ, Buchman A, et al:
Late-onset MNGIE due to partial loss of thymidine phosphorylase
activity. Ann Neurol. 58:649–652. 2005.PubMed/NCBI
|
|
16
|
Massa R, Tessa A, Margollicci M, et al:
Late-onset MNGIE without peripheral neuropathy due to incomplete
loss of thymidine phosphorylase activity. Neuromuscul Disord.
19:837–840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Libernini L, Lupis C, Mastrangelo M, et
al: Mitochondrial neurogastrointestinal encephalomyopathy: novel
pathogenic mutations in thymidine phosphorylase gene in two Italian
brothers. Neuropediatrics. 43:201–208. 2012. View Article : Google Scholar
|
|
18
|
Ishikawa F, Miyazono K, Hellman U, et al:
Identification of angiogenic activity and the cloning and
expression of platelet-derived endothelial cell growth factor.
Nature. 338:557–562. 1989. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bakker JA, Schlesser P, Smeets HJ,
Francois B and Bierau J: Biochemical abnormalities in a patient
with thymidine phosphorylase deficiency with fatal outcome. J
Inherit Metab Dis. 2010:Feb 12–2010.(Epub ahead of print).
|
|
20
|
Haraguchi M, Tsujimoto H, Fukushima M, et
al: Targeted deletion of both thymidine phosphorylase and uridine
phosphorylase and consequent disorders in mice. Mol Cell Biol.
22:5212–5222. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
López LC, Akman HO, García-Cazorla A, et
al: Unbalanced deoxynucleotide pools cause mitochondrial DNA
instability in thymidine phosphorylase-deficient mice. Hum Mol
Genet. 18:714–722. 2009.PubMed/NCBI
|
|
22
|
Tamura K, Peterson D, Peterson N, Stecher
G, Nei M and Kumar S: MEGA5: molecular evolutionary genetics
analysis using maximum likelihood, evolutionary distance, and
maximum parsimony methods. Mol Biol Evol. 28:2731–2739. 2011.
View Article : Google Scholar
|
|
23
|
Ingman M and Gyllensten U: mtDB: Human
Mitochondrial Genome Database, a resource for population genetics
and medical sciences. Nucleic Acids Res. 34:D749–D751.
2006.PubMed/NCBI
|
|
24
|
Tanaka M, Takeyasu T, Fuku N, Li-Jun G and
Kurata M: Mitochondrial genome single nucleotide polymorphisms and
their phenotypes in the Japanese. Ann NY Acad Sci. 1011:7–20. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hagiwara K, Stenman G, Honda H, et al:
Organization and chromosomal localization of the human
platelet-derived endothelial cell growth factor gene. Mol Cell
Biol. 11:2125–2132. 1991.PubMed/NCBI
|
|
26
|
Asai K, Nakanishi K, Isobe I, et al:
Neurotrophic action of gliostatin on cortical neurons. Identity of
gliostatin and platelet-derived endothelial cell growth factor. J
Biol Chem. 267:20311–20316. 1992.PubMed/NCBI
|