Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignant tumors with high malignancy and mortality due to
lack of early diagnosis and its resistance to conventional
chemotherapy (1). HCC affects
approximately one million people every year worldwide, with the
incidence equal to the mortality. In 2008, HCC was listed as the
third most common cause of cancer-related mortality (2). Thus, early diagnosis is crucial in
order to increase the survival rate for patients (3). To date, alpha-fetoprotein (AFP)
together with imaging and pathology detection are commonly used in
early clinical diagnosis of liver cancer. However, the specificity
and sensitivity of AFP for liver cancer screening are not
satisfactory. With the development of molecular biology, a number
of new types of tumor markers have been discovered.
Golgi phosphoprotein 2 (Golph2) is a type II
Golgi-specific membrane protein that is predominantly expressed in
the epithelial cells of a number of human tissues (4). In normal human liver, Golph2 is only
expressed in biliary epithelial cells and is almost undetected in
liver cells. However, increased expression of Golph2 has been
reported to be correlated with numerous viral or non-viral
infectious liver diseases (5). It
was first identified in a search for upregulated hepatic genes in
acute giant-cell hepatitis (4)q
and then in patients with acute and chronic hepatitis (6). Studies revealed that Golph2 was
overexpressed in the serum of HCC patients (7,8). In
China, Mao et al first observed that the level of Golph2 in
the serum of patients with HCC infected by hepatitis B virus (HBV)
was significantly higher than HBV carriers, patients without
hepatic diseases and healthy adults (9). In addition, other studies reported
that the sensitivity of diagnosis of HCC by Golph2 (76%) was higher
than AFP (70%), indicating that Golph2 may be a novel and effective
serum biomarker for the diagnosis of HCC (10).
In the present study, we established hybridoma cell
lines that stably secrete anti-Golph2 monoclonal antibody (mAb).
Using selected, purified and enzyme-labeled anti-Golph2 mAb, we
detected the level of antigen in HCC and healthy samples by
double-antibody sandwich enzyme-linked immunosorbent assay
(s-ELISA) and expect this method to be used in diagnostics and
therapeutics in the future.
Materials and methods
Subjects
The study protocol was approved by the Central South
University’s Institutional Review Board, Changsha, Hunan, China and
written informed consent was obtained from each subject.
Demographic and clinical information was obtained, and a blood
sample was collected from each subject. All the subjects were
recruited from the clinics at Xiangya Second Hospital, Changsha,
Hunan between March 2012 and November 2012. The diagnosis of HCC
was made based on guidelines from the Chinese Society of
Hepatology, the Chinese Society of Infectious Diseases and the
Chinese Medical Association (11–13).
A 4 ml blood sample obtained from each subject prior to the
initiation of HCC treatment was centrifuged and the serum aliquoted
and stored at −80˚C until testing (n=30; age, 54.30±10.14 years;
HBV-positive). Control subjects were enrolled from staff with no
liver disease or risk factors for viral hepatitis, and who were in
normal physiological condition [n=30; age, 49.87±9.23 years; normal
control (NC)]. Balb/c mice were maintained according to
institutional animal care and use committee (IACUC-BIDMC)
protocols.
Expression, purification and
identification of recombinant protein
The recombinant plasmid pET21a(+)-TRX-Golph2, which
was constructed in the laboratory, was transformed into
Escherichia coli Rosetta (ATCC, Manassas, VA, USA) and
optimal expression of recombinant proteins was achieved through
controlling the concentration of
isopropyl-β-D-thiogalactopyranoside (IPTG) and growth conditions.
After induction, bacteria were harvested and centrifuged at 4˚C,
4,449 × g for 10 min, and the pellet was resuspended in 50 mmol/l
sodium phosphate with 0.3 mol/l NaCl (pH 8.0). The resuspended
cells were then lysed by sonication and centrifuged at 4°C, 10,012
× g for 10 min. The expression form of fusion protein TRX-Golph2
was analyzed by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE, 12% gel).
The recombinant protein TRX-Golph2 was further
purified by immobilized metal affinity chromatography (MagneHis™
Protein Purification System; Promega, Madison, WI, USA) under
native conditions. The purified protein was dialyzed in 1X
phosphate buffered saline (PBS) at 4°C overnight and condensed to a
high concentration.
The concentrated recombinant protein was separated
by SDS-PAGE on a 12% polyacrylamide gel and then transferred onto a
nitrocellulose membrane (Millipore, Billerica, MA, USA) by
electroblotting. Fat-free milk (5%) was used to block the membrane
at 37°C for 2 h. The membrane was then incubated with anti-His tag
antibody (1:2,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA) overnight in a 4°C refrigerator. The membrane was then washed
with PBS-Tween-20 (PBST) three times and incubated with the
horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary
antibody (1:1,000, Sigma, St. Louis, MO, USA) in a 37°C shaker for
1 h. After washing three times with PBST, the membrane was
visualized with an enhanced chemiluminescence (ECL) system (Pierce
Biotechnology, Inc., Rockford, IL, USA).
Immunization of mice
Eight-week-old Balb/c mice were immunized three
times with purified TRX-Golph2 fusion protein at days 0, 21 and 42
with 100, 50 and 50 μg, respectively, by peritoneal injection.
Complete and incomplete Freund’s adjuvant (1:1, Sigma) were used to
emulsify the protein in the first and other immunizations,
respectively. Ten days after the third immunization, mice were bled
from the caudal vein and the serum titer was detected by indirect
ELISA. Three days after the last booster immunization (100 μg), the
spleen of the mouse with the highest titer was removed under
sterile conditions to prepare a splenic lymphocyte suspension for
cell fusion.
Cell culture and fusion
Mouse myeloma cell line SP2/0 was cultured in 10%
fetal bovine serum (Hyclone, Tauranga, New Zealand) and RPMI-1640
medium (Gibco-BRL, Grand Island, NY, USA) in a 37°C, 5%
CO2 incubator. The feeder cells, obtained from the
peritoneal cavity of a normal BALB/c mouse by peritoneal injection
of RPMI-1640 medium, were plated in 96-well plates and cultured in
a 37°C, 5% CO2 incubator for 1 day prior to fusion.
Splenic lymphocytes (1×108) were fused with SP2/0 cells
(1×107) in 50% polyethylene glycol 1500 (PEG 1500,
Sigma-Aldrich, St. Louis, MO, USA). The hybridoma cells were
cultured in 96-well plates containing feeder cells and screened by
hypoxanthine-aminopterin-thymidine (HAT; Sigma-Aldrich) and
cultured in a 37°C, 5% CO2 incubator. Two weeks later,
indirect ELISA using purified TRX-Golph2 protein and HepG2 cells as
coating proteins, respectively, was performed to detect the
positive clones and then screened clones were recloned by limiting
dilution in hypoxanthine-thymidine (HT; Sigma-Aldrich) medium for a
further 2 weeks.
Screening of positive clones and
cloning
By observing the 96-well plates, we selected a
single clone to perform the indirect ELISA using purified
TRX-Golph2 protein and HepG2 cells as coating proteins,
respectively. The optical density (OD) was read at a wavelength of
405 nm (microplate reader, Tianshi, Beijing, China) and considered
positive when the ratio of OD test to OD control was greater than 3
times. The wells that were positive for both purified TRX-Golph2
protein and HepG2 cells were considered as positive wells and then
recloned 3–5 times by limiting dilution until their antibody
secretion was 100%. Finally, the positive clones in the 96-well
plates were transferred to 24-well plates in 10% fetal bovine serum
RPMI-1640 medium. The strain of hybridoma cells that secreted
specific mAb was established.
Preparation and titer determination of
anti-Golph2 mAb
Balb/c mice were injected with incomplete Freund’s
adjuvant (0.5 ml) by peritoneal injection. Three days later, the
mice were peritoneally injected with 5×106 hybridoma
cells diluted by D-Hank’s. Ten days later, ascitic fluid was
collected and centrifuged at 278 × g for 5 min to remove the
cellular deposition. Anti-Golph2 mAb in supernatant was diluted
into different gradients (1:100; 1:200; 1:400; 1:800; 1:1,600;
1:3,200; 1:6,400; 1:12,800; 1:25,600; 1:51,200) and added into
96-well plates coated with TRX-Golph2 antigen, respectively.
Indirect ELISA was performed to detect the titer of anti-Golph2
mAb. The OD was read at 405 nm.
Subclass determination and purification
of anti-Golph2 mAb
The subclass of anti-Golph2 mAb was determined using
the SBA Clonotyping System (SouthernBiotech, Birmingham, AL, USA)
according to the manufacturer’s instructions. The anti-Golph2 mAb
(3 ml) was added with equivalent precooled ammonium sulfate
overnight in a 4°C refrigerator. They were then centrifuged at 4°C,
1,738 × g for 15 min. The supernatant was decanted, solubilized by
adding PBS (0.02 M, 3 ml), and then filtered by Sephacryl S-300 HR
gel filtration (Pharmacia, USA). PBS containing 0.02%
NaN3 was used to wash the column and collected
components. The positive components confirmed by indirect ELISA
were dialyzed in PBS at 4°C overnight and condensed to a high
concentration. The purity was identified by SDS-PAGE (12%).
Determination of the relative mAb
affinity
A 96-well immunoplate was coated with the TRX-Golph2
fusion protein (4 μg/ml) at 4°C overnight, and then was blocked
with 2% fetal bovine serum at 37°C for 2 h. Serial dilutions of the
purified mAb were incubated at 37°C for 2 h. The plate was rinsed
and incubated with the HRP-conjugated goat anti-mouse antibody
(1:4,000 dilution) at 37°C for 1 h. After washing, TMB
(3,3′,5,5′-tetramethylbenzidine) substrate was used for color
development. The OD value was measured at 405 nm in order to
determine the relative affinity.
Western blot analysis of anti-Golph2 mAb
specificity
Total proteins extracted from HepG2 cells were
separated by SDS-PAGE on a 12% polyacrylamide gel and then
transferred onto a nitrocellulose membrane (Millipore) by
electroblotting. Fat-free milk (5%) was used to block the membrane
at 37°C for 2 h. The membrane was then incubated with purified
anti-Golph2 mAb (1:2,000) overnight in a 4°C refrigerator. The
membrane was then washed with PBS-Tween-20 (PBST) three times and
incubated with the HRP-conjugated goat anti-mouse secondary
antibody (1:1,000) in a 37°C shaker for 1 h. After washing three
times with PBST, the membrane was visualized with an ECL system
(Pierce Biotechnology, Inc.).
Immunocytochemistry
Appropriate amounts of cells were cultured on
microslides overnight. When the cells were adherent, slides were
fixed with 4% paraformaldehyde for 15 min. PBST (0.5%) was used to
wash the slides three times. Endogenous peroxidase activity was
blocked by 3% H2O2/methanol and non-specific
binding was blocked with 2% BSA-PBS. Diluted anti-Golph2 mAb (50
μl; 1:50) was added and incubated overnight in a 4°C refrigerator.
After three 0.5% PBST washes, the appropriate (1:200)
HRP-conjugated goat anti-mouse secondary antibody was added and
incubated at room temperature for 1 h. Slides were washed with 0.5%
PBST three times. Diaminobenzidine (DAB) was added and allowed to
react for 1 min. The reaction was stopped by adding water and
hematoxylin was added to counterstain for 2 min. Slides were
dehydrated with alcohol and mounted with neutral gummi.
Preparation of enzyme-labeled anti-Golph2
mAb
Modified sodium periodate oxidation was used for
labeling anti-Golph2 mAb (8A7B4) with high purity. The result and
the optimal working concentration of labeled anti-Golph2 mAb with
HRP were analyzed by direct ELISA.
Establishment and optimization of
double-antibody s-ELISA
A 96-well immunoplate was coated with purified
anti-Golph2 mAb (5C6D5, 20 μg/ml) at 4°C overnight and then blocked
with 2% BSA-PBS at 4°C overnight. After washing plates with PBST
three times, serum dilutions (1:2, 1:4, 1:8), strong positive (50
ng/ml antigen), weak positive (3 ng/ml antigen) and negative, were
incubated at 37°C for 2 h, respectively. The plates were washed
three times and enzyme-labeled anti-Golph2 mAb (1:300, 1:400,
1:500, 1:600) was added and the plates were incubated at 37°C for 1
h. The plates were washed again and colored with 100 μl
2,2-Azinobis (3-ethylbenzothiazoline-6-sulfonic acid) diammonium
salt (ABTS; Boehringer Mannheim GmbH, Germany) for 25 min at 37°C.
The OD was read at a wavelength of 405 nm. A chessboard titration
method was designed to examine the optimal working concentration of
HRP-labeled mAb and the dilution ratio of serum.
Analysis of serum samples by s-ELISA
The expression level of Golph2 in serum dilutions
was analyzed by s-ELISA.
Statistical analysis
All of the values which presented as the mean of
three paralleled individual experiments were entered into SPSS 13.0
software. The mean and standard deviation (SD) of the OD of
individual groups were calculated. The expression of Golph2 in
serum was described using box-and-whisker plots. The difference
between groups was analyzed using Student t-tests. P<0.05 was
considered to indicate a statistically significant result.
Results
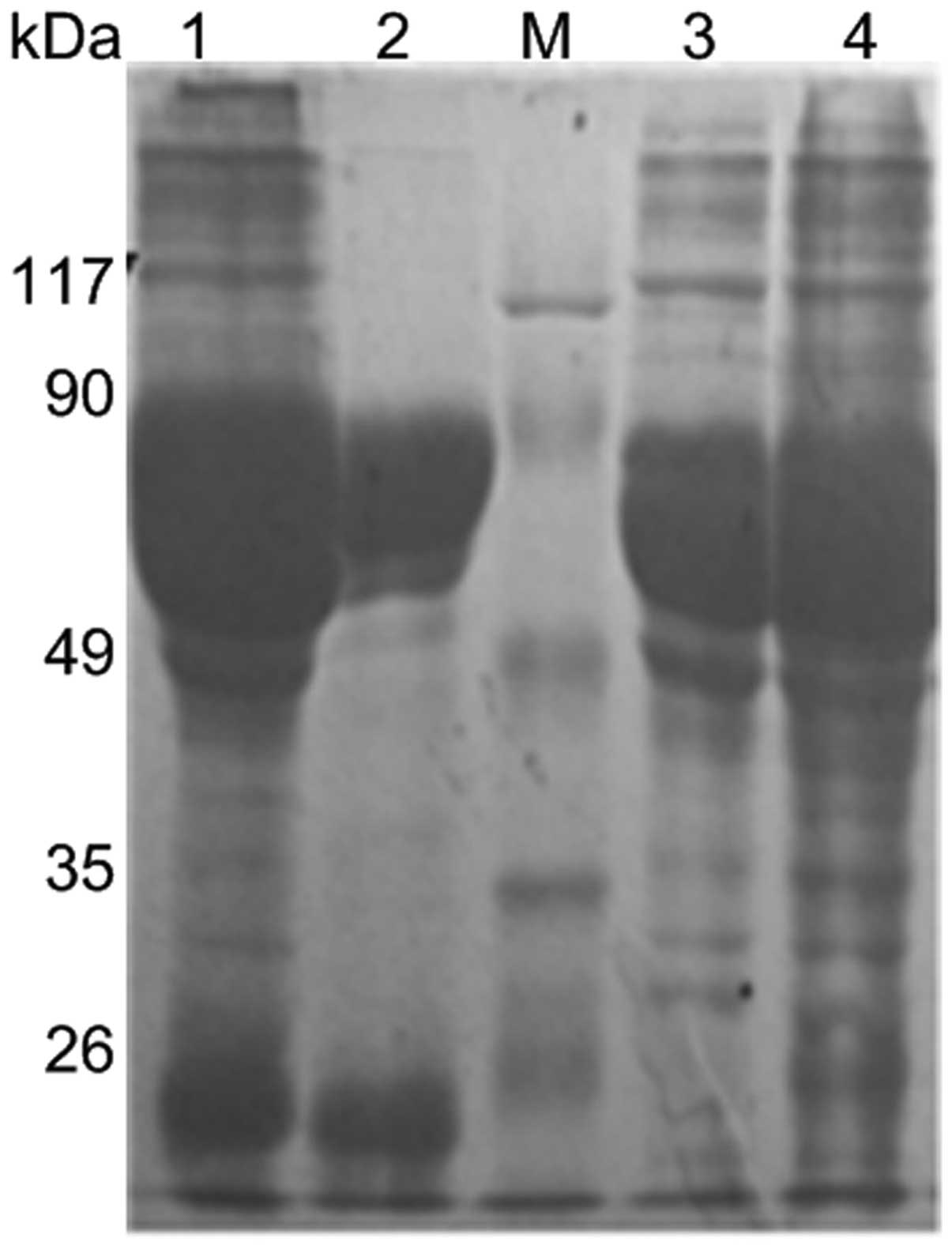
Expression, purification and
identification of recombinant protein
The recombinant fusion protein was expressed under
the optimizing prokaryotic expression conditions of 1 mM IPTG,
30°C, 2 × g, for 10 h and purified by immobilized metal affinity
chromatography under native conditions. The expressed and purified
protein were identified by 12% SDS-PAGE and subsequently stained
with Coomassie Brilliant Blue R250 (Fig. 1). The molecular weight of
TRX-Golph2 protein was ~73 kDa. The protein was then further
identified by western blot analysis using anti-His tag antibody
(Fig. 2).
Establishment of hybridoma cell lines and
preparation of anti-Golph2 mAb
Through the procedures of immunization, fusion and
clone selection, five hybridoma cell lines (5C6D5, 5B7F5, 7F5F3,
8A7B4, 8C9E8) that stably secreted anti-Golph2 mAb were obtained.
The identity, subclass and titer are shown in Table I.
 | Table ICharacterization of anti-Golph2
protein mAbs. |
Table I
Characterization of anti-Golph2
protein mAbs.
| Clone name | Titer | Subclass |
|---|
| 5B7F5 | 1:25600 | IgM (λ) |
| 5C6D5 | 1:51200 | IgM (κ) |
| 7F5F3 | 1:25600 | IgM (κ) |
| 8A7B4 | 1:12800 | IgG1 (κ) |
| 8C9E8 | 1:25600 | IgM (λ) |
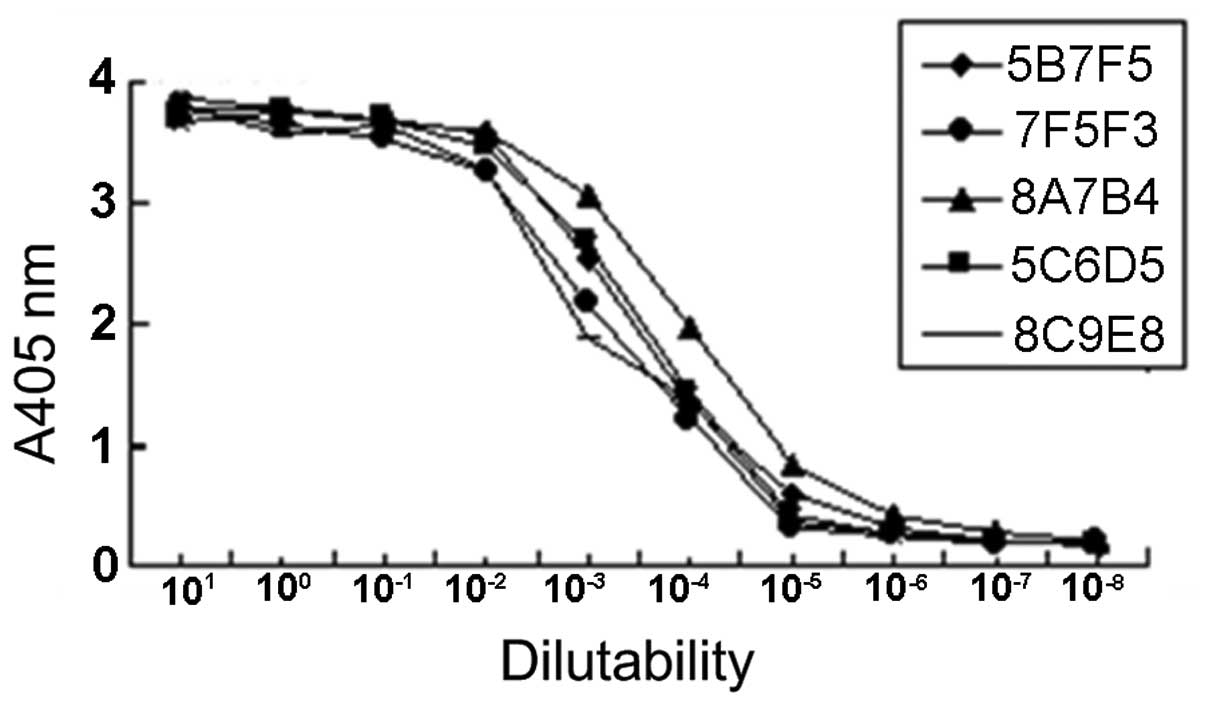
Investigation of mAb relative
affinity
The relative affinities of mAbs were defined by the
antibody concentration at which the OD value reached half the
maximal signal at the plateau stage of antigen-antibody binding.
The results revealed that the order of relative affinity of the
five selected mAbs was 8A7B4>5C6D5>5B7F5>8C9E8>7F5F3
(Fig. 3).
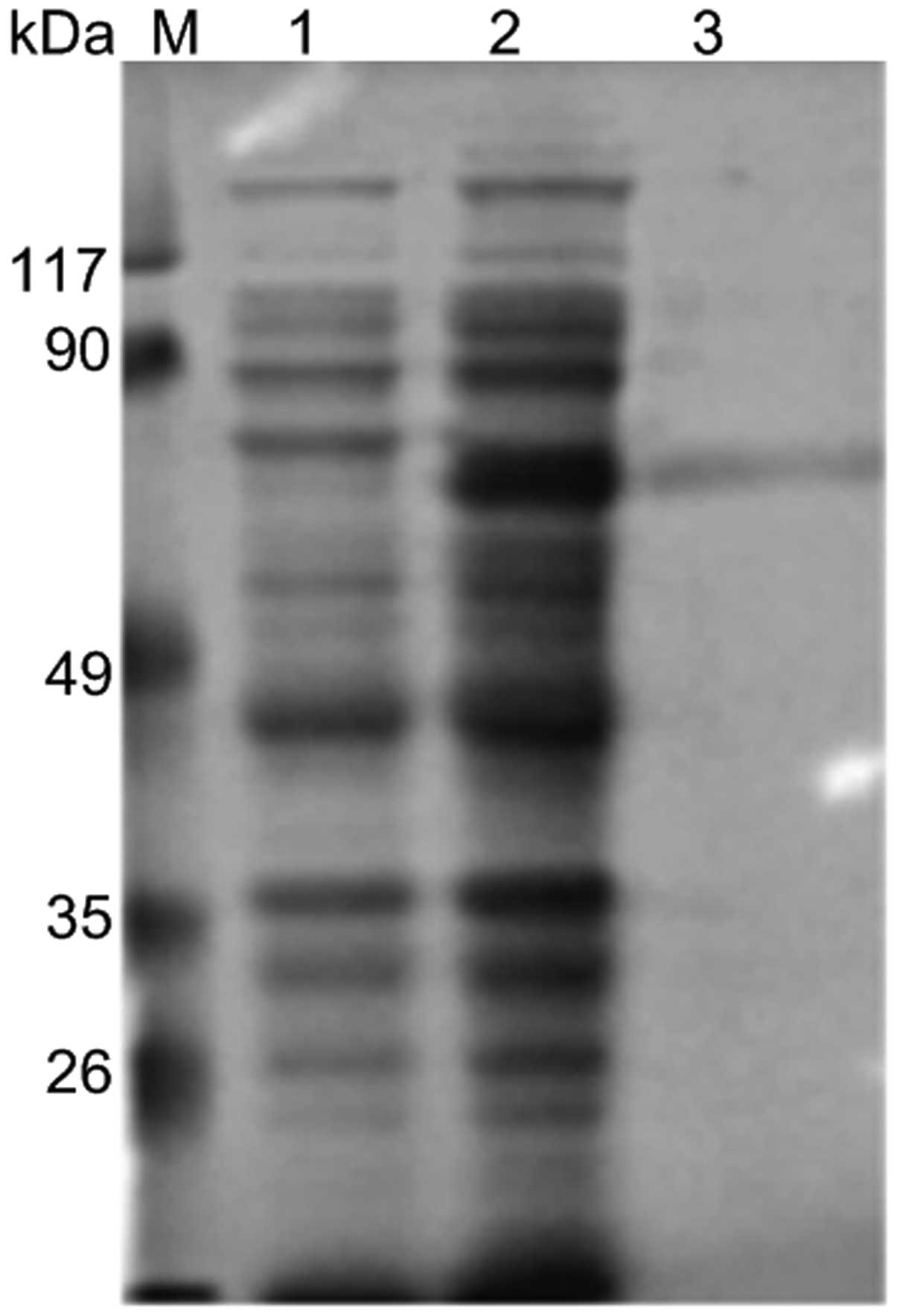
Purification of anti-Golph2 mAb
The anti-Golph2 mAb was purified by Sephacryl S-300
HR gel filtration. After dialyzing and condensing, the purity was
identified by SDS-PAGE (12%) (Fig.
4).
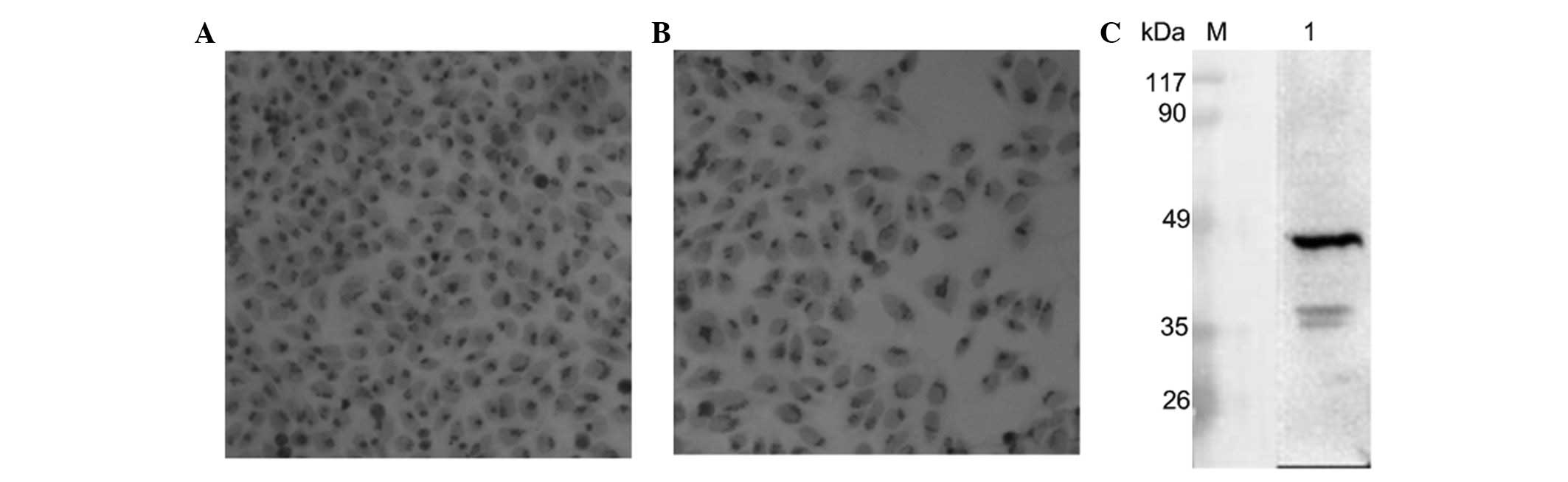
The specificity of anti-Golph2 mAb
Immunocytochemistry (HepG2) revealed that the
membrane of the Golgi was positively stained (Fig. 5A and B). By western blot analysis,
anti-Golph2 mAb (8A7B4) was shown to react specifically with Golph2
protein (45kDa) in accordance with the theoretical value (Fig. 5C).
Preparation of enzyme-labeled anti-Golph2
mAb
The anti-Golph2 mAb (8A7B4) was labeled with HRP by
modified sodium periodate oxidation. By direct ELISA, the working
concentration of labeled mAb was shown to be 1:500.
Establishment and optimization of
double-antibody s-ELISA
One serum sample from a known patient with
overexpression of Golph2 in serum was used for the determination of
the dilution ratio of serum. The optimal working concentration of
HRP-labeled mAb and the dilution ratio of serum were fixed on 1:500
and 1:2, respectively, by the chessboard titration method (Table II).
| Table IISelection of the optimal working
concentrations of HRP-marked mAb and the optimum serum
dilution. |
Table II
Selection of the optimal working
concentrations of HRP-marked mAb and the optimum serum
dilution.
| OD405 |
|---|
|
|
|---|
| Dilution of
HRP-marked mAb | Serum dilutions
(1:2) | Serum dilutions
(1:4) | Serum dilutions
(1:8) | Strong positive (50
ng/ml) | Weak positive (3
ng/ml) | Negative |
|---|
| 1:300 | 1.256 | 0.637 | 0.287 | 1.029 | 0.195 | 0.156 |
| 1:400 | 0.869 | 0.437 | 0.228 | 0.749 | 0.117 | 0.115 |
| 1:500 | 0.627 | 0.315 | 0.149 | 0.573 | 0.095 | 0.094 |
| 1:600 | 0.435 | 0.373 | 0.157 | 0.403 | 0.084 | 0.089 |
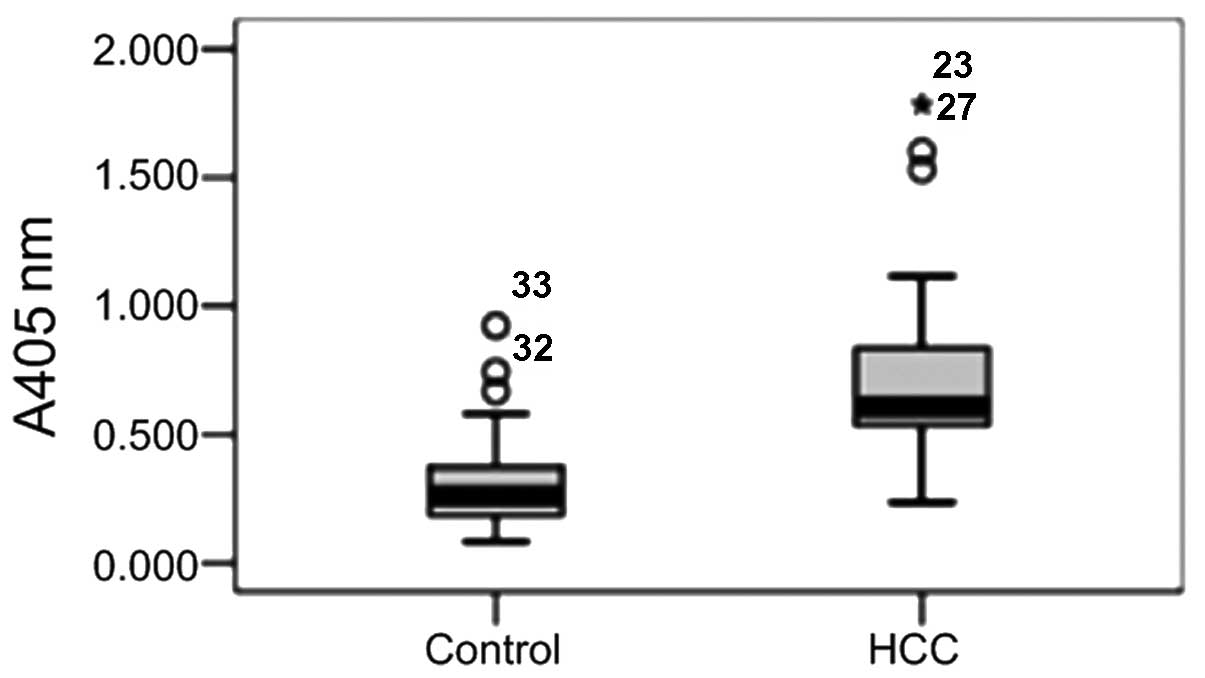
Detection of serum
The levels of antigen in the samples were detected
by s-ELISA using HRP-labeled anti-Golph2 mAb (Fig. 6). The median serum level of Golph2
in the HCC group significantly exceeded that of the healthy
controls (P<0.05). As shown in Fig.
6, the average serum content of Golph2 in HCC patients is
>100 μg/l, consistent with other studies using s-ELISA (8).
Discussion
Since it was first identified in acute giant-cell
hepatitis, tissue-based Golph2 overexpression has been confirmed in
HCC (8,14), lung adenocarcinoma (15), seminomas (16), renal cell cancer (17) and other malignant diseases.
Notably, a large number of studies have indicated that the serum
level of Golph2 may be a more reliable biomarker for the early
diagnosis of HCC than AFP. Previous studies have showed that Golph2
is an adenovirus-induced cellular protein whose expression is
regulated through E1A protein (4,18).
Golph2 overexpression, intracellular trafficking between the Golgi
and plasma membrane through an endosomal pathway, and the cleavage
of Golph2 may explain the secretion of Golph2 (19). However, the exact mechanism
involved remains unclear. mAbs against Golph2 prepared in this
study lay a foundation for further investigation of the interaction
between antigen epitopes and antibodies, and provide a tool for
determining the Golph2 secretion mechanism.
In our study, the E. coli system
Rosetta was selected to express Golph2 protein. When using a
prokaryotic system, it is difficult to express the full length of
Golph2. The Rosetta system contains rare codon tRNA that
improves protein expression, allowing Golph2 to be expressed. We
selected pET21a(+)-TRX as the prokaryotic expression vector. TRX is
a thioredoxin and is widely used in molecular cloning expression.
TRX protein prevents the target protein from being degraded by
endoproteinase and provides the ligand binding sites for affinity
purification. Furthermore, TRX contributes to folding of the target
protein and increases its soluble expression. The fusion protein
TRX-Golph2 can be expressed under native conditions. SDS-PAGE
showed that the fusion protein had a 78 kDa band. This is not in
accordance with the theoretical value, as Golph2 has abundant
acidic amino acids (PI=4.72), which causes the migration rate of
the recombinant protein to decline in the denatured SDS-PAGE
(20). After induction and
purification, we successfully obtained TRX-Golph2 in the soluble
form.
Using mAb technology with high purity and good
reproducibility (21,22), we selected five hybridoma cell
lines that stably secreted anti-Golph2 mAb. Two strains of
relatively high affinity mAb were selected and analyzed by western
blot analysis and immunocytochemistry. Western blotting showed that
the mAb recognized Golph2 protein specifically. Immunocytochemistry
revealed that Golph2 is located in the membrane of the Golgi. The
mAb with higher purity was labeled with HRP. The concentration of
the coated antibody was fixed at 20 μg/ml, and the optimal working
concentration of HRP-labeled mAb and the dilution ratio of serum
was confirmed to be 1:500 and 1:2, respectively, using the
chessboard titration method in s-ELISA.
Double-antibody s-ELISA is commonly used for testing
antigens. In the present study, HRP-labeled mAb with different
epitopes from coated mAb was produced for the first time. Previous
studies on Golph2 in serum used enzyme-labeled anti-Golph2
polyclonal antibody or a combination of anti-mAb with
enzyme-labeled anti-IgG to perform the double-antibody s-ELISA
(8). Polyclonal antibody has a
high level of cross reaction and can generate false positive
results, while mAb has high specificity and accuracy. Therefore,
our s-ELISA had significantly higher sensitivity and specificity
than the previous studies. Results showed that the expression level
of serum Golph2 in patients with HCC was markedly higher than that
in healthy individuals.
In conclusion, mAbs and s-ELISA against Golph2 may
be useful tools for the sensitive and specific diagnosis of HCC.
The preparation of humanized antibody has been published previously
(23), whereas the optimization of
humanized antibody requires further research. This study provides
the basis for optimization of humanized antibody.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (NSFC)-973 Project (2010CB833605).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics. CA Cancer J Clin. 62:10–29. 2012.
|
|
2
|
El-Serag HB, Marrero JA, Rudolph L and
Reddy KR: Diagnosis and treatment of hepatocellular carcinoma.
Gastroenterology. 134:1752–1763. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Trinchet JC, Alperovitch A, Bedossa P,
Degos F, Hainaut P and Beers BV: Epidemiology, prevention,
screening and diagnosis of hepatocellular carcinoma. Bull Cancer.
96:35–43. 2009.PubMed/NCBI
|
|
4
|
Kladney RD, Bulla GA, Guo L, et al: GP73,
a novel Golgi-localized protein upregulated by viral infection.
Gene. 249:53–65. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gu Y, Chen W, Zhao Y, Chen L and Peng T:
Quantitative analysis of elevated serum Golgi protein-73 expression
in patients with liver diseases. Ann Clin Biochem. 46:38–43. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iftikhar R, Kladney RD, Havlioglu N, et
al: Disease- and cell-specific expression of GP73 in human liver
disease. Am J Gastroenterol. 99:1087–1095. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Riener MO, Stenner F, Liewen H, et al:
Golgi phosphoprotein 2 (GOLPH2) expression in liver tumors and its
value as a serum marker in hepatocellular carcinomas. Hepatology.
49:1602–1609. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tian L, Wang Y, Xu D, et al: Serological
AFP/Golgi protein 73 could be a new diagnostic parameter of hepatic
diseases. Int J Cancer. 12:1923–1931. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mao Y, Yang H, Xu H, et al: Golgi protein
73 (GOLPH2) is a valuable serum marker for hepatocellular
carcinoma. Gut. 59:1687–1693. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou Y, Yin X, Ying J and Zhang B: Golgi
protein 73 versus alpha-fetoprotein as a biomarker for
hepatocellular carcinoma: a diagnostic meta-analysis. BMC Cancer.
12:172012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chinese Society of Hepatology.
2004.Guidelines for surgical treatment of primary hepatocellular
carcinoma. Zhonghua Gan Zang Bing Za Zhi. 13:329–330.
2005.PubMed/NCBI
|
|
12
|
Chinese Society of Infectious Diseases.
The guidelines of prevention and treatment for chronic hepatitis B.
Zhonghua Gan Zang Bing Za Zhi. 13:881–891. 2005.PubMed/NCBI
|
|
13
|
Chinese Medical Association. Guideline of
prevention and treatment of hepatitis C. Zhonghua Yu Fang Yi Xue Za
Zhi. 38:210–215. 2004.
|
|
14
|
Shi Y, Chen J, Li L, et al: A study of
diagnostic value of golgi protein gp73 and its genetic assay in
primary hepatic carcinoma. Technol Cancer Res Treat. 10:287–294.
2011.PubMed/NCBI
|
|
15
|
Zhang F, Gu Y, Li X, Wang W, He J and Peng
T: Up-regulated Golgi phosphoprotein 2 (GOLPH2) expression in lung
adenocarcinoma tissue. Clin Biochem. 43:983–991. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fritzsche FR, Kristiansen G, Riener MO,
Dietel M and Oelrich B: GOLPH2 expression may serve as diagnostic
marker in seminomas. BMC Urol. 10:42010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fritzsche FR, Riener MO, Dietel M, Moch H,
Jung K and Kristiansen G: GOLPH2 expression in renal cell cancer.
BMC Urol. 8:152008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kladney RD, Tollefson AE, Wold WS and
Fimmel CJ: Upregulation of the Golgi protein GP73 by adenovirus
infection requires the E1A CtBP interaction domain. Virology.
301:236–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hu L, Li L, Xie H, Gu Y and Peng T: The
Golgi localization of GOLPH2 (GP73/GOLM1) is determined by the
transmembrane and cytoplamic sequences. PLoS One. 6:e282072011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gong Y, Long Q, Xie H, Zhang T and Peng T:
Cloning and characterization of human Golgi phosphoprotein 2 gene
(GOLPH2/GP73/GOLM1) promoter. Biochem Biophys Res Commun.
421:713–720. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Köhler G and Milstein C: Continuous
cultures of fused cells secreting antibody of predefined
specificity. Nature. 256:495–497. 1975.
|
|
22
|
Gerdes J, Lemke H, Baisch H, et al: Cell
cycle analysis of a cell proliferation-associated human nuclear
antigen defined by the monoclonal antibody Ki-67. J Immunol.
133:1710–1715. 1984.PubMed/NCBI
|
|
23
|
Li F, Liu YH, Li YW, et al: Construction
and development of a mammalian cell-based full-length antibody
display library for targeting hepatocellular carcinoma. Appl
Microbiol Biotechnol. 96:1233–1241. 2012. View Article : Google Scholar : PubMed/NCBI
|