Introduction
Bone metabolism is predominantly regulated by
osteoblasts and osteoclasts, which are responsible for bone
formation and bone resorption, respectively (1). It is well known that osteoblasts
synthesize basic fibroblast growth factor (FGF-2), which is
embedded in the bone matrix (2).
During fracture repair, high levels of FGF-2 expression are
detected in osteoblasts (3). Data
suggest that FGF-2 is important in fracture healing, bone
remodeling and osteogenesis. There are four FGF receptor subtypes
with a high structural affinity (FGF receptors 1–4) (4) and it has been determined that FGF-2
activated the FGF receptors 1 and 2 in osteoblast-like MC3T3-E1
cells (5). It has also been
observed that FGF-2 stimulated the release of vascular endothelial
growth factor (VEGF), a mitogen that is highly specific for
vascular endothelial cells responding to various physiological
stimulants including insulin-like growth factor-1 and bone
morphogenetic protein (6). VEGF is
important in bone remodeling to stimulate angiogenesis inducing the
formation of bone microvasculature (7). Regarding the signaling mechanisms in
osteoblasts, it has been demonstrated that FGF-2-induced VEGF
release is positively regulated by p44/p42 mitogen-activated
protein (MAP) kinase and stress-activated protein
kinase/c-Jun N-terminal kinase (SAPK/JNK). However, it is
negatively regulated by p38 MAP kinase and p70 S6 kinase (8–10).
Heat shock proteins (HSPs) are induced by numerous
types of stress such as heat and chemical stress (11,12).
HSPs have been classified into groups including HSPA (HSP70), HSPB
(small HSPs), HSPC (HSP90) and HSPH (HSP110) (12). High-molecular-weight HSPs such as
HSPA (HSP70), HSPC (HSP90) and HSPH (HSP110) have been investigated
and were demonstrated to act as molecular chaperones that prevent
the aggregation of unfolded proteins and exert a cytoprotective
function (12). Small HSPs (HSPB)
with a monomer molecular mass of 12–43 kDa such as HSP27 (HSPB1),
αB-crystallin (HSPB5) and HSP20 (HSPB6), are constitutively
expressed in unstimulated cells and tissues such as skeletal,
smooth and cardiac muscle. Members of the HSPB group are currently
considered to be involved in essential functions such as protein
intracellular transport and the cytoskeletal architecture (12). It has been demonstrated that the
expression levels of HSP27 were low in unstimulated osteoblasts
(13). In addition, certain bone
modulating agents such as prostaglandins and transforming growth
factor-β are able to induce the expression of HSP27 via
intracellular signaling systems such as the activation of MAP
kinase in osteoblast-like MC3T3-E1 cells (13–18).
It has been observed that HSP27 is involved in regulating the
balance between the differentiation and apoptosis of osteoblasts
(19,20). However, the precise function of
HSPBs has yet to be elucidated, unlike that of the
high-molecular-weight HSPs.
It is generally observed that the functions of HSP27
(HSPB1) are regulated by post-translational modifications such as
phosphorylation (12). HSP27,
which normally exists as unphosphorylated oligomers (≤800 kDa), has
three phosphorylatable serine residues (Ser-15, Ser-78 and Ser-82).
When HSP27 is phosphorylated, a conformational change from the
aggregated form to the dimer occurs (21,22).
Prostaglandin D2 was observed to induce the phosphorylation of
HSP27 through p44/p42 MAP kinase and p38 MAP kinase in the MC3T3-E1
cells (23). In addition,
unphosphorylated HSP27 has a stimulatory effect on mineralization
whereas phosphorylated HSP27 enhances tumor necrosis
factor-α-stimulated interleukin-6 synthesis in these cells
(24,25). These results led to the hypothesis
that HSP27 is essential in the regulation of bone metabolism
through the control of osteoblast functions. However, the exact
role of HSP27 in bone metabolism remains to be fully elucidated. In
the present study, the involvement of HSP27 in the VEGF synthesis
stimulated by FGF-2 in osteoblast-like MC3T3-E1 cells was
investigated.
Materials and methods
Materials
The mouse VEGF enzyme-linked immunosorbent assay
(ELISA) kit and FGF-2 were purchased from R&D Systems, Inc.
(Minneapolis, MN, USA). Antibodies against phospho-specific p44/p42
MAP kinase, p44/p42 MAP kinase, phospho-specific p38 MAP kinase,
p38 MAP kinase, phospho-specific SAPK/JNK, SAPK/JNK,
phospho-specific p70 S6 kinase and p70 S6 kinase were obtained from
Cell Signaling, Technology, Inc. (Beverly, MA, USA). The enhanced
chemiluminescence (ECL) western blotting system was obtained from
GE Healthcare UK, Ltd. (Buckinghamshire, UK). The bicinchoninic
assay (BCA) Protein Assay Reagent kit was purchased from Pierce
Biotechnology, Inc., (Rockford, IL, USA). Other materials and
chemicals were obtained from commercial sources.
Cell culture
Cloned osteoblast-like MC3T3-E1 cells derived from
newborn mouse calvaria (26) were
maintained as previously described (27). Briefly, the cells were cultured in
α-minimum essential medium (α-MEM) containing 10% fetal calf serum
(FCS) at 37°C in a humidified atmosphere of 5% CO2 and
95% air.
Establishment of the cells stably
transfected with HSP27
Mutant-HSP27 plasmids, in which serine residues
(Ser-15, Ser-72 and Ser-82) were mutated to alanines to generate
constitutively non-phosphorylatable HSP27 (3A cells), or were
mutated to aspartic acid to generate constitutively
phospho-mimicking HSP27 (3D cells), were provided by Dr C. Schafer
(Klinikum Großhadern, Ludwig-Maximilians University, Munich,
Germany). For stable transfections, MC3T3-E1 cells
(5×105 cells) were cultured in 6-well plates. The cells
were transfected with 2 μg of the wild-type (WT) or the mutant
HSP27 plasmids expressing geneticin (G418; EMD Chemicals, Inc., San
Diego, CA, USA) resistance using 12 μl of the UniFECTOR
transfection reagent (B-Bridge International, Cupertino, CA, USA)
in 1 ml α-MEM medium without FCS. Medium (1 ml) with 10% FCS was
added 5 h following transfection. The cells were incubated in the
presence of 400 μg/ml of G418. After 2 weeks, single G418-resistant
colonies were obtained by serial dilution in 96-well plates. The
colonies were maintained and analyzed individually for the
expression of HSP27.
Western blot analysis
The cultured cells were stimulated with 30 ng/ml
FGF-2 in α-MEM containing 0.3% FCS for the indicated periods. The
cells were washed twice with phosphate-buffered saline, lysed,
homogenized and sonicated in a lysis buffer containing 62.5 mM
Tris/HCl (pH 6.8), 2% sodium dodecyl sulfate (SDS) and 10%
glycerol. SDS-polyacrylamide gel electrophoresis (PAGE) was
conducted according to Laemmli (28) on 10% polyacrylamide gels. A western
blot analysis was performed as previously described (13) using antibodies against
phospho-specific p44/p42 MAP kinase, p44/p42 MAP kinase,
phospho-specific p38 MAP kinase, p38 MAP kinase, phospho-specific
SAPK/JNK, SAPK/JNK, phospho-specific p70 S6 kinase and p70 S6
kinase with peroxidase-labeled antibodies as the secondary
antibodies. The peroxidase activity on the polyvinylidene fluoride
(PVDF) membrane was visualized on X-ray film by the ECL western
blotting system.
VEGF assay
The cultured cells were stimulated by 30 and 50
ng/ml of FGF-2 in 1 ml α-MEM containing 0.3% FCS for the indicated
periods. The conditioned medium and cell lysates were collected at
the end of the incubation period. The VEGF concentration was
measured by a VEGF ELISA kit. The absorbance of the ELISA samples
was measured at 450 and 560 nm with an EL 340 Microplate Reader
BioKinetic plate (Bio-Tek Instruments, Inc., Winooski, VT, USA).
The levels of VEGF release were adjusted for the respective whole
cell lysates. The protein levels of the cells were measured by a
BCA Protein Assay Reagent kit according to the manufacturer’s
instructions.
Statistical analysis
The data were analyzed by analysis of variance
(ANOVA) followed by the Bonferroni method for multiple comparisons
between pairs. P<0.05 was considered to indicate a statistically
significant difference. Data are presented as the mean ± SEM of
triplicate independent determinations.
Results
Effect of overexpressed HSP27 on
FGF-2-stimulated VEGF release in MC3T3-E1 cells
It has previously been demonstrated that FGF-2
stimulated VEGF release in osteoblast-like MC3T3-E1 cells (8). In order to investigate the
involvement of HSP27 on the FGF-2-induced VEGF release, its effect
was investigated in WT HSP27 cDNA-transfected MC3T3-E1 cells. As
previously observed (24), the
‘empty’ cells, which were transfected with an empty vector,
represented the normal parental MC3T3-E1 cells and the WT cells
represented the MC3T3-E1 cells overexpressing the wild type-HSP27.
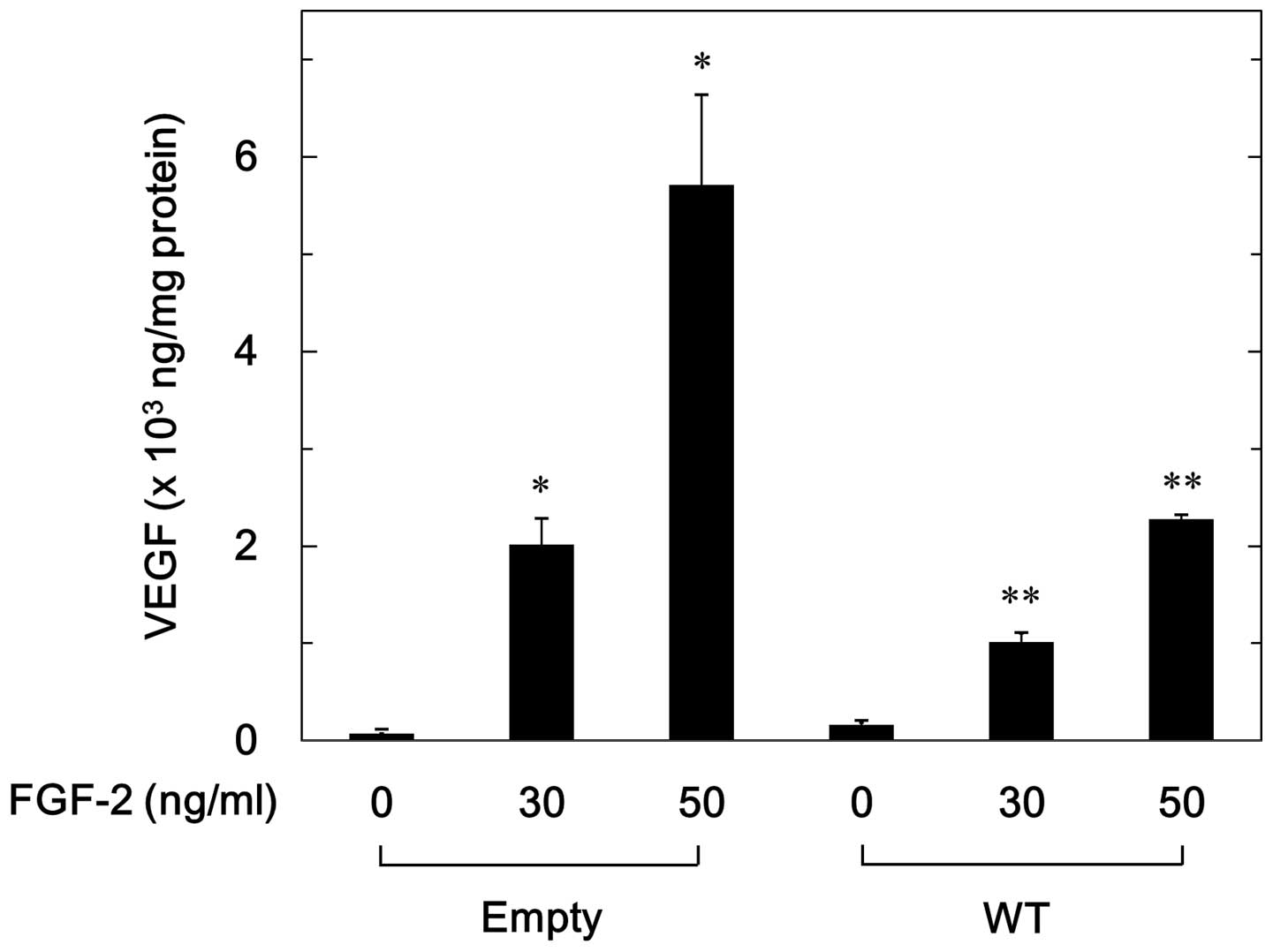
FGF-2 (30–50 ng/ml) significantly stimulated VEGF release in a
dose-dependent manner in the WT and empty cells (Fig. 1). The levels of VEGF release
induced by FGF-2 were markedly lower in the WT cells compared with
the empty cells (Fig. 1). The
levels of VEGF induced by FGF-2 (50 ng/ml) in the WT cells were
~40% lower compared with those in the empty cells.
Effects of FGF-2 on intracellular
signaling in the WT HSP27-transfected MC3T3-E1 cells and the empty
vector-transfected MC3T3-E1 cells
In a previous study it was demonstrated that VEGF
release stimulated by FGF-2 was positively regulated by p44/p42 MAP
kinase and SAPK/JNK but negatively regulated by p38 MAP kinase and
p70 S6 kinase in osteoblast-like MC3T3-E1 cells (8–10).
The effects of FGF-2 on the intracellular signaling in the WT and
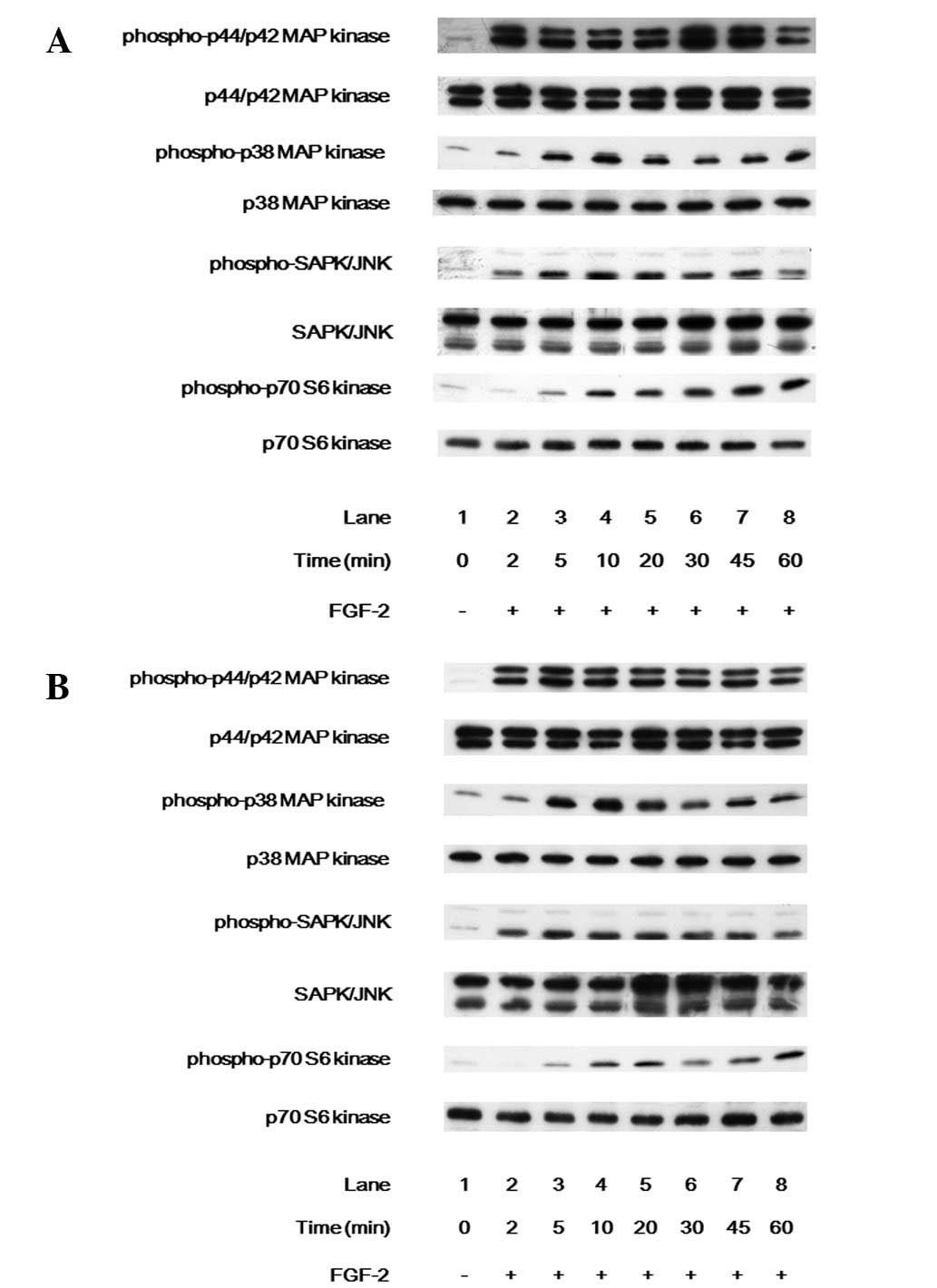
empty cells were observed. FGF-2 markedly induced the
phosphorylation of p44/p42 MAP kinase, p38 MAP kinase, SAPK/JNK and
p70 S6 kinase in the WT (Fig. 2A)
and empty cells (Fig. 2B).
However, no significant differences were noted in the
phosphorylation of p44/p42 MAP kinase, p38 MAP kinase, SAPK/JNK or
p70 S6 kinase between the WT and empty cells.
Effect of phosphorylated HSP27 on
FGF-2-stimulated VEGF release in MC3T3-E1 cells
To elucidate whether the effect of FGF-2 on VEGF
release is affected by the phosphorylation status of HSP27 in
osteoblast-like MC3T3-E1 cells, the effects of FGF-2 on VEGF
release in the 3D cells compared with that in the 3A cells was
observed. As previously demonstrated (24), HSP27 was overexpressed in the 3A
and 3D cells, and the phosphorylation levels of HSP27 (Ser-82) in
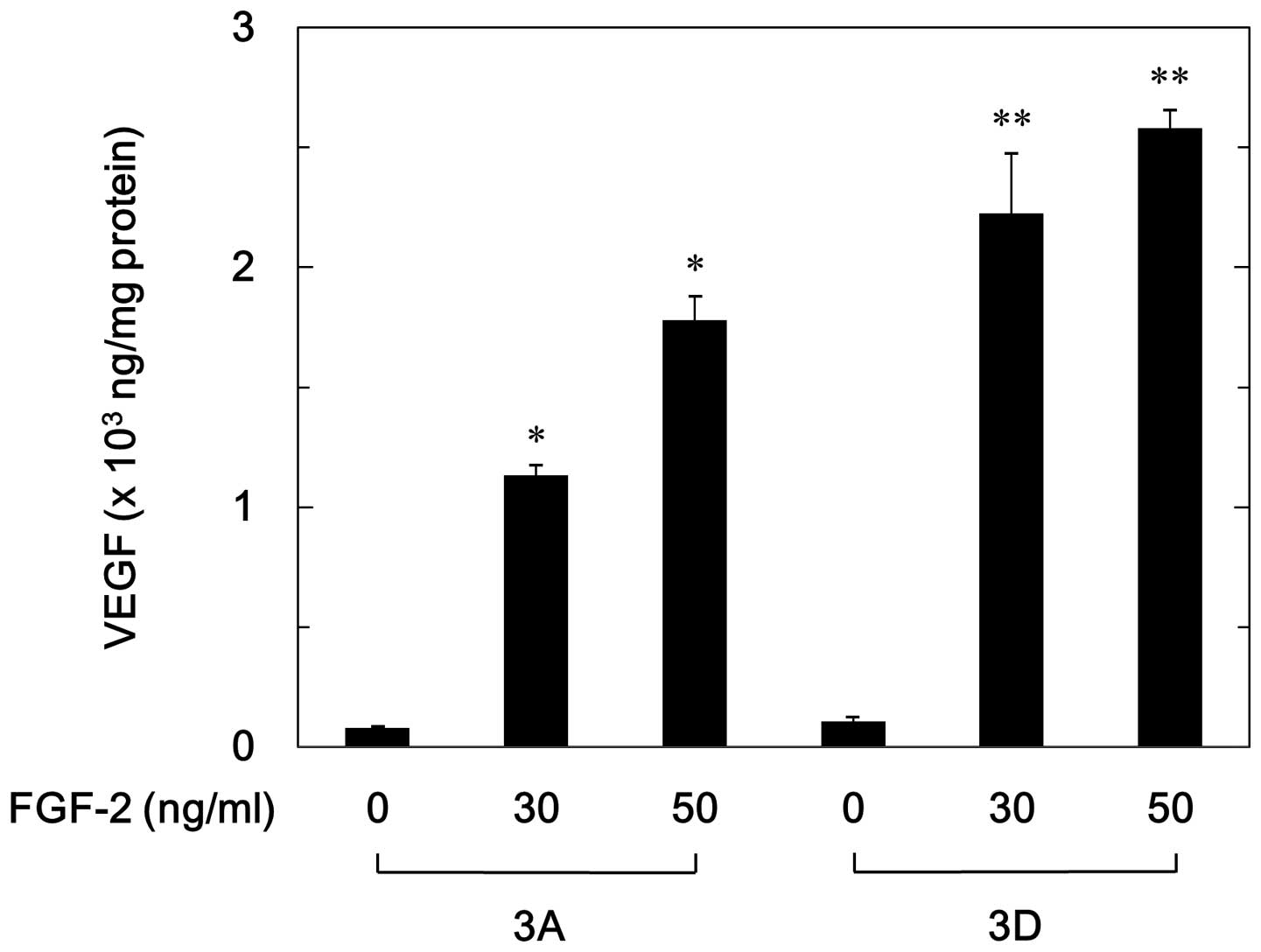
the 3D cells were much greater than those in the 3A cells. FGF-2
significantly stimulated VEGF release in a dose-dependent manner
with a range of 30–50 ng/ml in the 3A and 3D cells (Fig. 3). However, the levels of VEGF
released in the 3D cells were significantly higher than those in
the 3A cells (Fig. 3). The levels
of VEGF induced by FGF-2 (30 ng/ml) in the 3A cells were ~50% lower
compared with those in the 3D cells.
Effects of phosphorylated HSP27 on the
intracellular signaling of FGF-2 in MC3T3-E1 cells
The effects of FGF-2 on the intracellular signaling
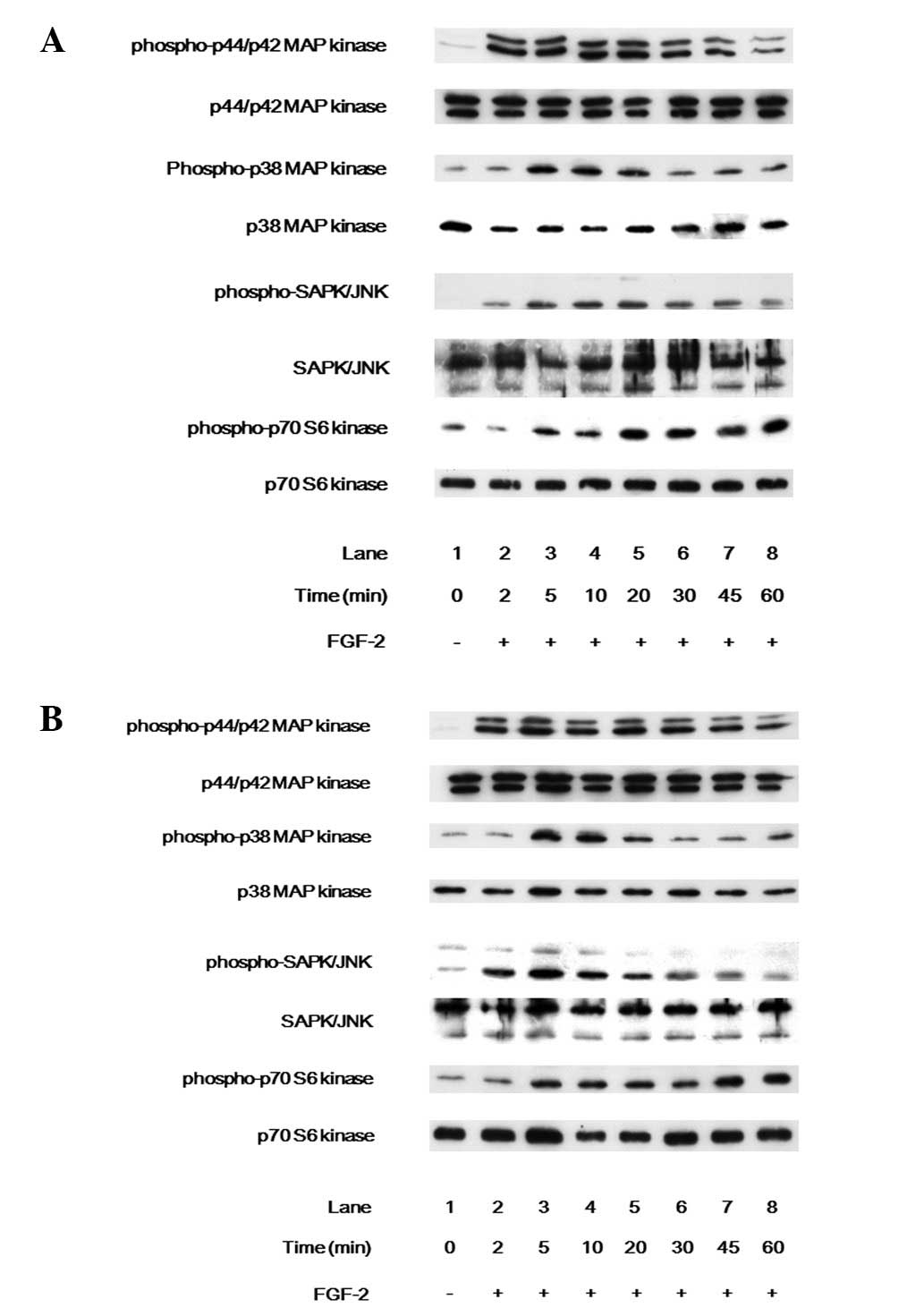
in the 3A and 3D cells were investigated. FGF-2 markedly induced
the phosphorylation of p44/p42 MAP kinase, p38 MAP kinase, SAPK/JNK
and p70 S6 kinase in the 3A (Fig.
4A) and 3D (Fig. 4B) cells.
However, no significant differences were noted in the
phosphorylation of p44/p42 MAP kinase, p38 MAP kinase, SAPK/JNK or
p70 S6 kinase between the 3A and 3D cells.
Discussion
In the present study, the effect of HSP27 on
FGF-2-stimulated VEGF synthesis in osteoblast-like MC3T3-E1 cells
was observed. The levels of FGF-2-stimulated VEGF release were
significantly attenuated in the HSP27-overexpressing MC3T3-E1 cells
compared with those in the empty vector-transfected MC3T3-E1 cells.
It was previously demonstrated that the levels of HSP27 were low in
the unstimulated-MC3T3-E1 cells and that the expression of HSP27 is
not significant in the empty cells (13,24).
The results of these studies suggested that HSP27 exhibited an
inhibitory effect on the FGF-2-induced VEGF synthesis in
osteoblast-like MC3T3-E1 cells.
In a resting state, HSP27 exists in an aggregated
form (≤800 kDa), which is phosphorylated at three serine residues
(Ser-15, Ser-78 and Ser 82). The phosphorylation is accompanied by
a conformational change from the aggregated form to the dissociated
dimer form (21). The effects of
the phosphorylation status of HSP27 on FGF-2-stimulated VEGF
synthesis using two types of mutant HSP27-transfected cells were
demonstrated. The 3A cells overexpressed non-phosphorylatable HSP27
and the 3D cells overexpressed mutant-HSP27, mimicking the
phosphorylated protein (24). The
levels of FGF-2-stimulated VEGF release were significantly lower in
the 3A cells compared with the 3D cells. In a previous study
(24), it was observed that HSP27
was overexpressed in the WT cells but was not phosphorylated.
Therefore it is likely that unphosphorylated HSP27 suppresses the
FGF-2-induced VEGF synthesis in osteoblast-like MC3T3-E1 cells. In
addition, the phosphorylation status of HSP27 may be able to induce
the VEGF synthesis in osteoblasts.
Concerning the regulatory mechanism underlying
FGF-2-stimulated VEGF synthesis in osteoblasts, it was previously
observed that the FGF-2-stimulated VEGF release was positively
regulated by the activation of p44/p42 MAP kinase and SAPK/JNK
(8,9). However, it was negatively regulated
by the activation of p38 MAP kinase and p70 S6 kinase in
osteoblast-like MC3T3-E1 cells (8–10).
In the present study, to investigate the effect of HSP27 expression
on the activation of p44/p42 MAP kinase, p38 MAP kinase, SAPK/JNK
and p70 S6 kinase, the FGF-2-induced phosphorylation levels of
these intracellular signaling molecules in the HSP27-transfected
MC3T3-E1 cells was also determined. However, the phosphorylation
levels of these molecules were not significantly different among
the four types of transfected MC3T3-E1 cells (empty vector, WT
HSP27, non-phosphorylatable HSP27, and phospho-mimic HSP27).
Therefore, it appears unlikely that HSP27 inhibited the
FGF-2-stimulated VEGF synthesis through the activation of p44/p42
MAP kinase, p38 MAP kinase, SAPK/JNK and p70 S6 kinase, or at a
point upstream of these molecules. Phosphorylated HSP27 was able to
change its localization from the cytoplasm to the perinuclear area
and acted as a functional regulator of the endoplasmic reticulum,
contributing to the regulation of osteocalcin synthesis (24). Thus, it is probable that the
localization change of HSP27 due to phosphorylation attenuated the
inhibitory activity of HSP27 in the VEGF synthesis induced by FGF-2
in osteoblasts.
Osteoporosis is a predominant clinical problem in
developed countries. The pathology of osteoporosis is a reduction
of the bone mineral density, which is a risk factor for bone
fractures (29). An increase in
FGF-2 expression in osteoblasts is detected during fracture repair.
In addition, VEGF induces the angiogenesis of the microvasculature
in the bone tissue which is important in bone remodeling (3,6).
Moreover, it has been observed that VEGF regulated the balance
between osteoblast and adipocyte differentiation (30). The regulation of VEGF-related
mechanisms, therefore, are predicted to determine novel aspects of
bone remodeling adjustment. Thus, the results demonstrating the
involvement of HSP27 in osteoblast function may provide a novel
therapeutic target for bone metabolic diseases such as
osteoporosis. Further investigation is required to determine the
precise mechanism of action of HSP27 in osteoblasts and in bone
metabolism.
In conclusion, the results suggest that
unphosphorylated HSP27 exerts an inhibitory effect on
FGF-2-stimulated VEGF synthesis in osteoblasts.
Acknowledgements
The authors would like to thank Dr C. Schafer for
providing the mutant HSP27 cDNA and Yumiko Kurokawa for technical
assistance. This study was supported by a Grant-in-Aid for
Scientific Research (grant no. 19591042) from the Ministry of
Education, Science, Sports and Culture of Japan, the Foundation for
Growth Science and the Research Funding for Longevity Sciences
(grant nos. 22-4 and 23-9) from the National Center for Geriatrics
and Gerontology, Japan.
References
|
1
|
Karsenty G and Wagner EF: Reaching a
genetic and molecular understanding of skeletal development. Dev
Cell. 2:389–406. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mirams M, Robinson BG, Mason RS and Nelson
AE: Bone as a source of FGF23: regulation by phosphate? Bone.
35:1192–1199. 2004.PubMed/NCBI
|
|
3
|
Barnes GL, Kostenuik PJ, Gerstenfeld LC
and Einhorn TA: Growth factor regulation of fracture repair. J Bone
Miner Res. 14:1805–1815. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Itoh N and Ornitz DM: Evolution of the Fgf
and Fgfr gene families. Trends Genet. 20:563–569. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Suzuki A, Shinoda J, Kanda S, Oiso Y and
Kozawa O: Basic fibroblast growth factor stimulates
phosphatidylcholine-hydrolyzing phospholipase D in osteoblast-like
cells. J Cell Biochem. 63:491–499. 1996. View Article : Google Scholar
|
|
6
|
Zelzer E and Olsen BR: Multiple roles of
vascular endothelial growth factor (VEGF) in skeletal development,
growth, and repair. Curr Top Dev Biol. 65:169–187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schipani E, Maes C, Carmeliet G and
Semenza GL: Regulation of osteogenesis-angiogenesis coupling by
HIFs and VEGF. J Bone Miner Res. 24:1347–1353. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tokuda H, Kozawa O and Uematsu T: Basic
fibroblast growth factor stimulates vascular endothelial growth
factor release in osteoblasts: divergent regulation by p42/p44
mitogen-activated protein kinase and p38 mitogen-activated protein
kinase. J Bone Miner Res. 15:2371–2379. 2000. View Article : Google Scholar
|
|
9
|
Tokuda H, Hirade K, Wang X, Oiso Y and
Kozawa O: Involvement of SAPK/JNK in basic fibroblast growth
factor-induced vascular endothelial growth factor release in
osteoblasts. J Endocrinol. 177:101–107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takai S, Tokuda H, Hanai Y, Harada A,
Yasuda E, Matsushima-Nishiwaki R, Kato H, Ogura S, Ohta T and
Kozawa O: Negative regulation by p70 S6 kinase of FGF-2-stimulated
VEGF release through stress-activated protein kinase/c-Jun
N-terminal kinase in osteoblasts. J Bone Miner Res. 22:337–346.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Taylor RP and Benjamin IJ: Small heat
shock proteins: a new classification scheme in mammals. J Mol Cell
Cardiol. 38:433–444. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mymrikov EV, Seit-Nebi AS and Gusev NB:
Large potentials of small heat shock proteins. Physiol Rev.
91:1123–1159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kozawa O, Niwa M, Matsuno H, Ishisaki A,
Kato K and Uematsu T: Stimulatory effect of basic fibroblast growth
factor on induction of heat shock protein 27 in osteoblasts: role
of protein kinase C. Arch Biochem Biophys. 388:237–242. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kozawa O, Otsuka T, Hatakeyama D, Niwa M,
Matsuno H, Ito H, Kato K, Matsui N and Uematsu T: Mechanism of
prostaglandin D(2)-stimulated heat shock protein 27 induction in
osteoblasts. Cell Signal. 13:535–541. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hatakeyama D, Kozawa O, Niwa M, Matsuno H,
Kato K, Tatematsu N, Shibata T and Uematsu T: Inhibition by
adenylyl cyclase-cAMP system of ET-1-induced HSP27 in osteoblasts.
Am J Physiol Endocrinol Metab. 281:E1260–E1266. 2001.PubMed/NCBI
|
|
16
|
Tokuda H, Kozawa O, Niwa M, Matsuno H,
Kato K and Uematsu T: Mechanism of prostaglandin E2-stimulated heat
shock protein 27 induction in osteoblast-like MC3T3-E1 cells. J
Endocrinol. 172:271–281. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tokuda H, Niwa M, Ishisaki A, Nakajima K,
Ito H, Kato K and Kozawa O: Involvement of stress-activated protein
kinase (SAPK)/c-Jun N-terminal kinase (JNK) in prostaglandin
F2alpha-induced heat shock protein 27 in osteoblasts.
Prostaglandins Leukot Essent Fatty Acids. 70:441–447. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hayashi K, Takai S, Matsushima-Nishiwaki
R, Hanai Y, Kato K, Tokuda H and Kozawa O: (−)-Epigallocatechin
gallate reduces transforming growth factor beta-stimulated HSP27
induction through the suppression of stress-activated protein
kinase/c-Jun N-terminal kinase in osteoblasts. Life Sci.
82:1012–1017. 2008.
|
|
19
|
Tiffee JC, Griffin JP and Cooper LF:
Immunolocalization of stress proteins and extracellular matrix
proteins in the rat tibia. Tissue Cell. 32:141–147. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leonardi R, Barbato E, Paganelli C and Lo
Muzio L: Immunolocalization of heat shock protein 27 in developing
jaw bones and tooth germs of human fetuses. Calcif Tissue Int.
75:509–516. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kostenko S and Moens U: Heat shock protein
27 phosphorylation: kinases, phosphatases, functions and pathology.
Cell Mol Life Sci. 66:3289–3307. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kostenko S, Johannessen M and Moens U:
PKA-induced F-actin rearrangement requires phosphorylation of Hsp27
by the MAPKAP kinase MK5. Cell Signal. 21:712–718. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takai S, Tokuda H, Yoshida M, Yasuda E,
Matsushima-Nishiwaki R, Harada A, Kato K and Kozawa O:
Prostaglandin D2 induces the phosphorylation of HSP27 in
osteoblasts: function of the MAP kinase superfamily. Prostaglandins
Leukot Essent Fatty Acids. 75:61–67. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kato K, Adachi S, Matsushima-Nishiwaki R,
Minamitani C, Natsume H, Katagiri Y, Hirose Y, Mizutani J, Tokuda
H, Kozawa O and Otsuka T: Regulation by heat shock protein 27 of
osteocalcin synthesis in osteoblasts. Endocrinology. 152:1872–1882.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kato K, Tokuda H, Mizutani J, Adachi S,
Matsushima-Nishiwaki R, Natsume H, Kozawa O and Otsuka T: Role of
HSP27 in tumor necrosis factor-α-stimulated interleukin-6 synthesis
in osteoblasts. Int J Mol Med. 28:887–893. 2011.
|
|
26
|
Sudo H, Kodama HA, Amagai Y, Yamamoto S
and Kasai S: In vitro differentiation and calcification in a new
clonal osteogenic cell line derived from newborn mouse calvaria. J
Cell Biol. 96:191–198. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kozawa O, Suzuki A, Tokuda H and Uematsu
T: Prostaglandin F2alpha stimulates interleukin-6 synthesis via
activation of PKC in osteoblast-like cells. Am J Physiol.
272:E208–E211. 1997.PubMed/NCBI
|
|
28
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Unnanuntana A, Gladnick BP, Donnelly E and
Lane JM: The assessment of fracture risk. J Bone Joint Surg Am.
92:743–753. 2010. View Article : Google Scholar
|
|
30
|
Liu Y, Beredsen AD, Jia S, Lotunum S,
Baron R, Ferrara N and Olsen BR: Intracellular VEGF regulates the
balance between osteoblast and adipocyte differentiation. J Clin
Invest. 122:3101–3113. 2012. View
Article : Google Scholar : PubMed/NCBI
|