Introduction
Hepatitis C virus (HCV) is a major public health
concern worldwide. Approximately 170 million people suffer from
chronic HCV and are at risk of developing cirrhosis and
hepatocellular carcinoma. In Pakistan alone, 10 million people are
infected with HCV and 50% of them are infected with the 3a subtype.
HCV is a small hepatotropic virus, and member of the
Flaviviridae family, infecting 170–180 million people
worldwide (1). Globally, 0.25–1.25
million new cases of HCV infection have been reported per year
(2). The core protein is the first
structural protein encoded by the HCV open reading frame (ORF),
consisting of 191 amino acids in its immature form. It is one of
the potential targets for specific drugs against HCV, as it is well
conserved in all HCV genotypes and interacts with a number of
cellular factors of the host immune system (3,4).
Expression of the HCV core protein results in suppression of type I
interferon (IFN) signaling leading to the reduction of
phosphorylated STAT1 (P-STAT1). HCV core protein and STAT1 are
reported to have a direct interaction involving residues in the
N-terminal portion of the HCV core (amino acids 1–23) (5). Mutations in the N-terminal of the
core protein are expected to modulate antiviral response in
general, as well as response to conventional pegylated interferon
(PEG-IFN) and ribavirin (PEG-IFN/ribavirin) combination therapy,
eventually leading to sustained virological response.
Emerging HCV resistance to the current standard
available treatment, PEG-IFN/ribavirin combination therapy, is of
great concern, as is its low response and toxicity (6). Although new direct acting antivirals
(DAAs) targeting HCV NS3–4A protease, namely telaprevir and
boceprevir, have shown an increase in the sustained virological
response (SVR) of up to 70% in patients infected with HCV genotype
1 (7), the conventional
PEG-IFN/ribavirin treatment remains part of the therapy. Clinical
studies have suggested that non-synonymous mutations are induced by
ribavirin monotherapy and thus increase IFN sensitivity (8). The SVR has been shown to be markedly
augmented by the addition of ribavirin to IFN monotherapy, with an
increase in the response rate and a reduction in the relapse rate
being observed (9). Mathematical
model applications have revealed viremic decay following
combination therapy (10).
In this study we propose a possible mechanism of
PEG-IFN/ribavirin-induced SVR. We suggest that PEG-IFN/ribavirin
therapy-induced amino acid changes in the N-terminus of the HCV
core are associated with viral clearance or persistence.
Materials and methods
Patient demographics
Patients with a positive PCR test for HCV, confirmed
by Atta-ur-Rahman School of Applied Biosciences (ASAB) Diagnostics,
were enrolled for this study under the approval of the Internal
Review Board (IRB) of ASAB, National University of Sciences and
Technology, Pakistan and a patient consent form was duly signed for
each patient. All patients were infected with genotype 3a, the most
prevalent genotype in Pakistan. The genotype of the patients was
determined using the method described by Ohno et al(11). The HCV patients selected for this
study were from two different groups. The patients in group A were
receiving treatment with pegylated interferon α-2a (PEG-IFN α2a)
180 μg/week and ribavirin 800 mg/day for 24 weeks. In group B,
patients that were recently diagnosed and had viral titer and
alanine transaminase (ALT) levels relatively close to those of
group A were selected (Table I).
The follow-up for the treated patients was carried out for 24 weeks
following the completion of treatment. The viral loads and ALT
levels of the patients were measured at 12-week intervals (Table I). Viral RNA was quantified using a
Bio-Rad RoboGene HCV amplification kit (Bio-Rad, Hercules, CA,
USA), whereas Microlab 300 (Merck, Germany) was employed for ALT
measurements. Sequencing of the isolated virus core gene was
performed after 12 weeks of PEG-IFN/ribavirin bitherapy. To compare
the mutations induced by ribavirin, the HCV core gene was also
cloned and sequenced from HCV-infected patients without liver
complications and who had not received any treatment.
 | Table IDemographic and follow-up data of
hepatitis C virus (HCV) infected patients. |
Table I
Demographic and follow-up data of
hepatitis C virus (HCV) infected patients.
| Patients undergoing
interferon/ribavirin treatment | Untreated
patients |
|---|
|
|
|---|
| Patient code | Age (Yrs) | Gender | Base line viral
titer/ALT | 12-week viral
titer/ALT | 24-week viral
titer/ALT | 36-week viral
titer/ALT | 48-week viral
titer/ALT | Patient code | Age (Yrs) | Gender | Viral titer/ml ALT
level U/l |
|---|
| PT1 | 42 | Female |
5×109/110 |
2×105/58 |
3×105/80 | - | - | P1 | 32 | Female |
5.6×106/58 |
| PT2 | 40 | Male |
5×108/110 |
3×105/56 |
3×105/80 | - | - | P2 | 44 | Female |
1×106/98 |
| PT3 | 39 | Female |
7×106/96 |
2×105/49 | <4000/48 | <4000/43 | <4000/43 | P3 | 40 | Male |
5.6×106/62 |
| PT4 | 45 | Male |
9×107/127 |
4×104/50 | <4000/50 | <4000/46 | <4000/46 | P4 | 37 | Male |
1.7×105/78 |
| PT5 | 41 | Male |
4×108/90 |
4×104/45 | <4000/42 | <4000/40 | <4000/40 | P5 | 29 | Male |
8×106/90 |
| PT6 | 39 | Female |
8×107/98 |
7×104/47 | <4000/47 | <4000/38 | <4000/38 | P6 | 32 | Female |
5×106/68 |
| PT7 | 40 | Male |
6×108/94 |
4×106/72 | <4000/48 | <4000/36 | <4000/42 | P7 | 37 | Male |
9×104/98 |
| PT8 | 43 | Male |
9×106/105 |
9×105/68 | <4000/48 | <4000/42 | <4000/38 | P8 | 37 | Female |
2×106/127 |
PCR amplification cloning and sequencing
of HCV core gene
For PCR amplification of the core gene, viral RNA
was extracted from patient serum by using an RNA extraction kit
(Qiagen, Hamburg, Germany) according to the manufacturer's
instructions. The primers used for cDNA synthesis and PCR
amplification were: 5′-AAA GAA TTC GCC ACC ATG CTA GAG TGG CGG AAT
ACG TCT GGC C-3′ (sense) and 5′-CCC GCG GCC GCT TAA CTG GCT GCT GGA
TGA ATT AAG C-3′ (antisense). Purified PCR products were cloned in
PCRII TOPO Cloning vector (Invitrogen, Singapore) as instructed by
the manufacturer. Two clones from each patient were subjected to
sequencing using a CEQ 8000 genetic analysis system (Beckman
Coulter, Miami, FL, USA) as described previously (12). Sequences from the present patients
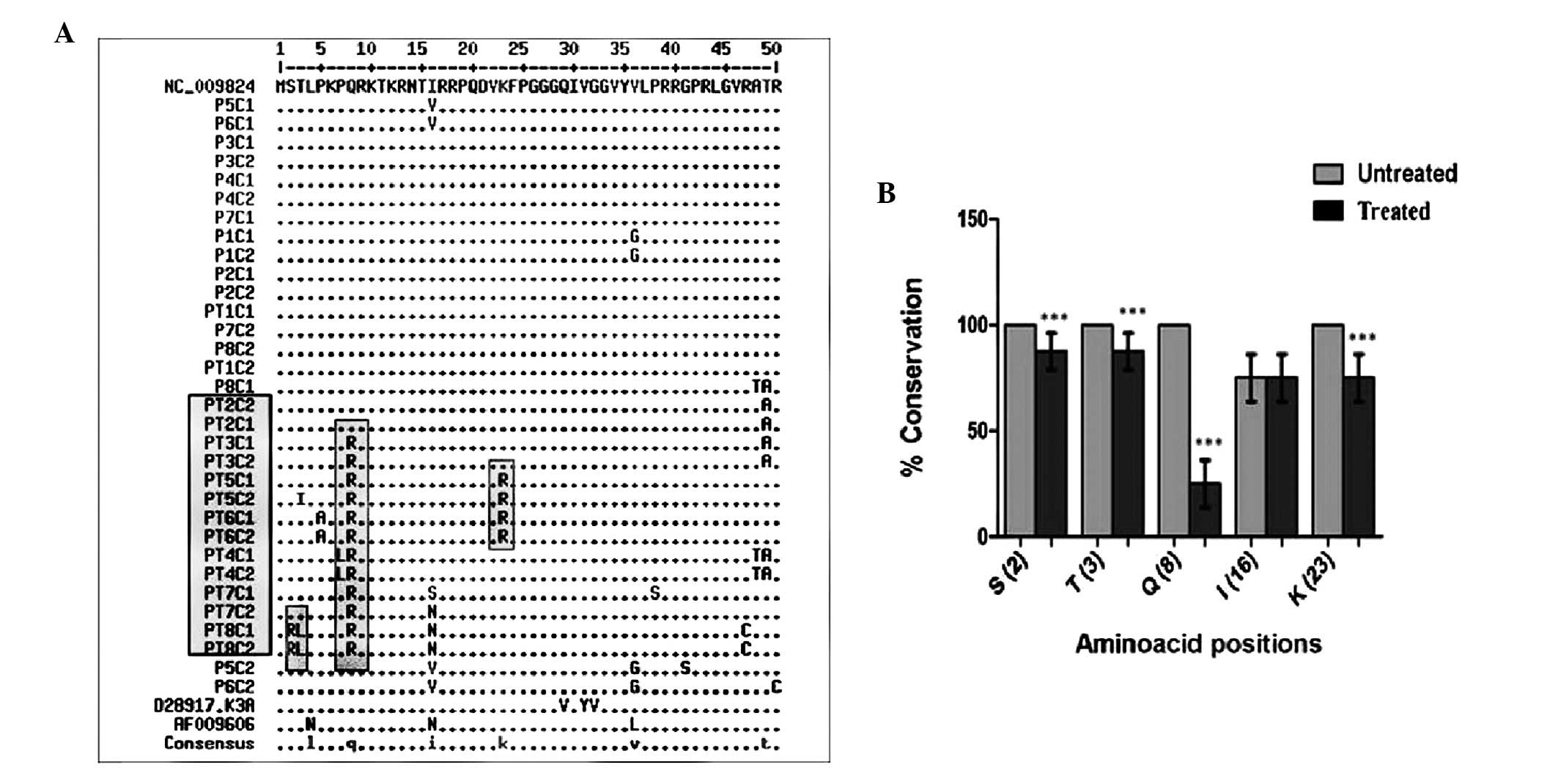
were aligned with reference isolates (Fig. 1). The aligned sequences of HCV core
(>3000 sequences) from the European database (http://euhcvdb.ibcp.fr/euHCVdb/) were analyzed to
check the conservation of the residues involved in core-STAT1
interactions in the N-terminus of the core gene.
Molecular modeling of HCV core gene and
its in silico interaction with STAT1
The HCV core gene consensus sequence of all 16
clones from the untreated patients was submitted to the I-TASSER
online web server (13) for
molecular modeling and the model with the highest C-value was
selected for further analysis. The model was refined with energy
minimization by subjecting it to ionized water box and
physiological concentrations. The AMBER 99 force field was used to
minimize its energy after protonation of the system by fixing its
charges and lone pairs. The minimized model was extracted from the
solvent system and was docked with the STAT1 protein (pdb id 1yvl).
The protein interaction between core and STAT1 was studied using
the HADDOCK web server (14).
Based on previous studies, residues 1–23 of the core were selected
as the active site and residues 577–684 of STAT1 were selected as
passive residues for this interaction. Different contact types,
including ionic cutoff 4.5, hydrophobic cutoff 4.5, hydrogen bonds
and disulfide cutoff 2.5, were evaluated between the core and STAT1
using the default bond angles used in the Molecular Operating
Environment (MOE). The sequence separation used was 4 residues
apart. Histidine was selected as it is basic in character whereas
methionine was characterized as hydrophobic in nature. The
mutations observed at core residues 2, 3, 8, 16 and 23 (Fig. 1) were incorporated into the in
silico interaction model. In order to investigate the further
significance of the observed mutations in the core
STAT1-interacting domain (amino acids 2, 3, 8, 16, 23) two way
ANOVA was applied on mutational data from treated and untreated
core residues. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
Sequence analysis
For sequence comparison, H77 genotype 1a was taken
as a reference strain for amino acid positioning. NZL1 and K3a
isolates of GT3a were taken as references for sequence analysis
(Fig. 1). Notably, as compared
with reference isolates and clones from untreated patients, few
major differences were observed in the N-terminal region of the
core. Residues 2, 3, 8, 16 and 23 were frequently mutated in
treated patients as compared with untreated patients (Fig. 1; Table II) and significant differences
(P<0.001) were recorded at positions 2, 3, 8 and 23.
Comprehensive analysis of the aligned core sequences reported in
the European database showed that residues 2, 3, 8 and 23 are well
conserved across all genotypes (Table III). Position 16 was not well
conserved and the mutations observed had no effect on core-STAT1
interactions.
| Table IIIn vivo mutations observed in
the hepatitis C virus core proteins from treated patients and their
effects on STAT1 interaction. |
Table II
In vivo mutations observed in
the hepatitis C virus core proteins from treated patients and their
effects on STAT1 interaction.
| Core residues | Mutations | Contact
alteration |
|---|
| 2 | S/R | No contacts
observed |
| 3 | T/L,I | No contacts
observed |
| 7 | P/L | Contact established
with Val 642 and Ile 647 of STAT1 |
| 8 | R/Q | No contacts
observed |
| 16 | S/N,I | Contact conserved
either N/I with Asp 627 |
| 23 | K/R | No contact
observed |
| Table IIIVariability and contact report of
hepatitis C virus core residues involved in the core-STAT1
interaction. |
Table III
Variability and contact report of
hepatitis C virus core residues involved in the core-STAT1
interaction.
| Position in
core | Actual residue | Mutated
residues | % variability | Contact report
core-STAT1 interaction |
|---|
| 2 | S | N=1/3498 | - | No contact
observed |
| | G=1/3498 | - | No contact
observed |
| 3 | T | A=3 | A=0.001 | No contact
observed |
| | R=2 | | No contact
observed |
| | M=1 | | No contact
observed |
| 7 | P | R=1 | L=0.001 | No contact
observed |
| | L=4 | - | Hydrophobic bonding
between leucine 7 to valine 642 and isoleucine 647 |
| 8 | Q | | | Hydrogen bonding
between arginine at position 8 and asparagine 646 |
| | R=7 | R=0.002 | No contact
observed |
| | P=11 | P=0.003 | No contact
observed |
| | H=2 | P=0.001 | Hydrogen bonding
between histidine 8 to tyrosine 634 |
| | K=2 | P=0.001 | Hydrogen bonding
between lysine 8 to asparagine 650. |
| | S=1 | P=0.001 | No contact
observed |
| 23 | K | R=10 | R=0.002 | No contact
observed |
| | E=4 | | No contact
observed |
| | S=31 | | No contact
observed |
In silico characterization of the STAT1
binding domain of the core protein
Sequence variations observed in the present study
were mostly found in the N-terminus of the protein, a region which
has been previously shown to interfere with IFN signaling by
interacting with the STAT1-SH2 domain (15). We therefore investigated how these
amino acid changes potentially affect core-STAT1 interactions. For
this purpose, an in silico approach based on prediction of
molecular docking was used. Although the structure of STAT1 is
known, the HCV core protein structure has not been reported. We
therefore started by determining a structural model for the HCV
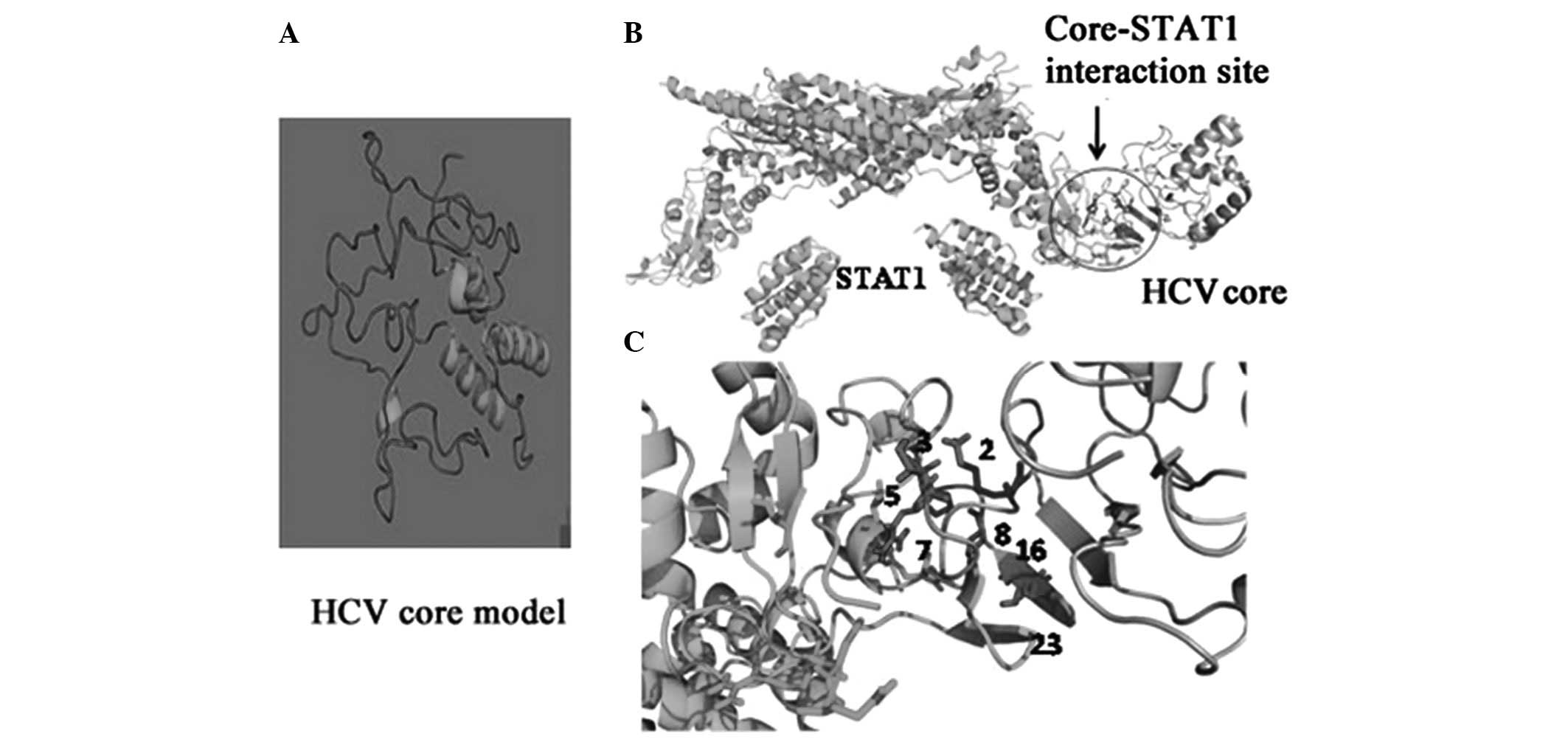
core protein (Fig. 2A). The
characterization of interaction contacts between the HCV core and
STAT1 were then determined (Fig.
2B; Table II) and the contact
details of the interacting residues are provided in Table III. Based on our molecular
modeling approaches, amino acids S, T, Q and K at positions 2, 3, 8
and 23, respectively, appear critical for core-STAT1 interaction.
Changes in these residues, as observed in some of our clones,
resulted in loss of contact between core and STAT1 (Fig. 2; Tables II and III). The follow-up data and the
core-STAT1 docking results clearly correlate the SVR observed in
six out of eight patients that carried observed mutations.
Follow-up of the untreated patients was not conducted, as they were
recommended for treatment.
Discussion
The mechanism of viral persistence and clearance has
not been well elucidated. Viral capsid proteins have been proposed
previously as targets for anti-viral drugs, as they are well
conserved across the 6 major genotypes (16). In the current study, the HCV core
gene was amplified from the serum of patients that were undergoing
PEG-IFN/ribavirin treatment for 12 weeks and from treatment-naïve
patients. As the core quasispecies tends to be conserved during
acute HCV infection (17), in the
present study patients without liver complications and at a
relatively early stage of disease were enrolled and thus a more
conserved core gene was anticipated.
Notably, as compared with isolates from untreated
patients (Fig. 1), few significant
differences were observed in the N-terminal region of the cores
from treated patients. Comprehensive analysis of the aligned core
sequences reported in the European database showed that residues 2,
3, 8 and 23 are well conserved across all genotypes. Amino acid
changes in this part of the protein are known to modulate viral
assembly or core interactions with host factors (18). The N-terminal region of the core
(amino acids 1–23) has been shown to block IFN signaling by
interaction with the STAT1-SH2 domain that plays a significant role
in HCV resistance to IFN therapy (19). In silico molecular docking
was used to observe the potential effects of these changes on the
core-STAT1 interaction. For this purpose, the HCV core protein
structure was modeled (Fig. 2A)
and the interaction contacts between the HCV core and STAT1 were
determined (Fig. 2B). The contact
details of the interacting residues are provided in Table II. Based on our molecular modeling
approaches, amino acids S, T, Q and K at residues 2, 3, 8 and 23
appear critical for core-STAT1 interaction. Changes in these
residues, as observed in the majority of our clones from treated
patients, resulted in a loss of contact between the core and STAT1.
Mutations at similar positions were rarely reported in the HCV
database and these residues tend to be conserved among various
genotypes.
Follow-up information (Table I) revealed that the core mutations
observed in six patients at critical residues resulted in a loss of
contact with STAT1, thus ensuring better antiviral response and
facilitating viral clearance. However, two of the patients,
patients 1 (PT1) and 2 (PT2), showed no mutation at these
positions. These two patients were non-responders and discontinued
therapy after six months (Table
I). Notably, in patient 4 (PT4), the virus had counteracted the
loss of the STAT1 interaction at position 8 by a P>L shift at
core position 7 that resulted in the establishment of a new
interaction with Val 642/Ile 647 of STAT1 (Table III). This new contact may
modulate STAT1 signaling and a relapse may occur following the
accumulation of the resistant variant. An early virological
response reported for genotype 3a was not evident in the current
study, possibly due to the small sample size; however, the
identification of non-responders is not unusual for genotype 3a. We
have recently recorded a significant difference in the mutation
rate of HCV glycoprotein E2 in treated vs. untreated patients and
have observed for the first time a glycosylation position shift in
envelope protein E2 that results in antibody escape variants,
giving the virus a chance to survive following the therapeutic
response (unpublished data).
Previous reports have indicated that amino acid
substitutions at position 70 and/or 91 in the HCV core protein
region of patients infected with HCV-1b are pretreatment predictors
of a poor virological response to PEG-IFN/ribavirin combination
therapy and telaprevir/PEG-IFN/ribavirin triple therapy (20,21).
In all patients included in this study, the core position 70 was
occupied by arginine as reported for genotype 1b and should favor
the treatment response, however the failure of patients 1 and 2 to
respond to treatment suggest that there may be more than one
predictor of therapeutic outcomes. Core residue 91 appears to be
genotype-specific and thus may contribute to the genotype-specific
antiviral response to IFN/ribavirin therapy. Another study,
however, described that the ribavirin monotherapy-induced mutagenic
effect, studied in the context of the NS3 and NS5B regions of HCV,
was reduced in patients receiving PEG-IFN and ribavirin combination
therapy, possibly due to the antiviral action of IFN (22). In the current study, since the
ribavirin monotherapy was not included due to its absence from the
general medical practices prevailing in Pakistan, there is a
possibility that certain other error mutations may have been
immediately eliminated by the concurrently administered IFN. This
may account for the relatively small number of mutations observed
in the current study, despite the presence of a mutagenic analog in
the combination bitherapy.
In conclusion, this study suggests that
IFN/ribavirin bitherapy-induced mutations in the STAT1-interacting
domain of the HCV core protein may be responsible for the improved
therapeutic response and viral clearance, at least in the GT3a
genotype, the most prevalent genotype in Pakistan. However, this
treatment may give rise to resistant variants that are able to
escape the current therapy. In addition, this study indicates for
the first time that residues 2, 3, 8, and 23 of the HCV core are
critical for the core-STAT1 interaction and we propose these
residues as a potential target for antiviral drug design.
Acknowledgements
The authors acknowledge Dr Jean Dubuisson for his
help in writing and improving this manuscript and ASAB Diagnostics
for HCV patient enrollment and for the follow-up records. This
study constitutes partial fulfillment for the degree of Doctor of
Philosophy for Anjum S. from ASAB (former NCVI), National
University of Science and Technology, Islamabad, Pakistan. The
authors also acknowledge HEC Pakistan and French split PhD
fellowship (EGIDE) for supporting this study.
References
|
1
|
Alter MJ, Mast EE, Moyer LA and Margolis
HS: Hepatitis C. Infect Dis Clin North Am. 12:13–26. 1998.
View Article : Google Scholar
|
|
2
|
Chen SL and Morgan TR: The natural history
of hepatitis C virus (HCV) infection. Int J Med Sci. 3:47–52.
1997.
|
|
3
|
Lin W, Kim SS, Yeung E, Kamegaya Y,
Blackard JT, Kim KA, Holtzman MJ and Chung RT: Hepatitis C virus
core protein blocks interferon signaling by interaction with the
STAT1 SH2 domain. J Virol. 80:9226–9235. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Melén K, Fagerlund R, Nyqvist M, Keskinen
P and Julkunen I: Expression of hepatitis C virus core protein
inhibits interferon-induced nuclear import of STATs. J Med Virol.
73:536–547. 2004.PubMed/NCBI
|
|
5
|
de Lucas S, Bartolome J and Carreno V:
Hepatitis C virus core protein down-regulates transcription of
interferon-induced antiviral genes. J Infect Dis. 191:93–99.
2005.PubMed/NCBI
|
|
6
|
Hadziyannis S, Sette H Jr, Morgan TR,
Balan V, Diago M, Marcellin P, Ramadori G, Bodenheimer H Jr,
Bernstein D, Rizzetto M, Zeuzem S, Pockros PJ, Lin A and Ackrill
AM; PEGASYS International Study Group. Peginterferon-α2a and
ribavirin therapy in chronic hepatitis C: A randomized study of
treatment duration and ribavirin dose. Annal Intern Med.
140:346–355. 2004.
|
|
7
|
Dore GJ, Matthews GV and Rockstroh J:
Future of hepatitis C therapy: development of direct-acting
antivirals. Curr Opin HIV AIDS. 6:508–513. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Asahina Y, Izumi N, Enomoto N, Uchihara M,
Kurosaki M, Onuki Y, Nishimura Y, Ueda K, Tsuchiya K, Nakanishi H,
Kitamura T and Miyake S: Mutagenic effects of ribavirin and
response to interferon/ribavirin combination therapy in chronic
hepatitis C. J Hepatol. 43:623–629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chung RT, Gale M Jr, Polyak SJ, Lemon SM,
Liang TJ and Hoofnagle JH: Mechanisms of action of interferon and
ribavirin in chronic hepatitis C: Summary of a workshop.
Hepatology. 47:306–320. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Herrmann E, Lee JH, Marinos G, Modi M and
Zeuzem S: Effect of ribavirin on hepatitis C viral kinetics in
patients treated with pegylated interferon. Hepatology.
37:1351–1358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohno T, Mizokami M, Wu RR, Saleh MG, Ohba
KI, Orito E, Mukaide M, Williams R and Lau JY: New hepatitis C
virus (HCV) genotyping system that allows for identification of HCV
genotypes 1a, 1b, 2a, 2b, 3a, 3b, 4, 5a, and 6a. J Clin Microbiol.
35:201–207. 1997.PubMed/NCBI
|
|
12
|
Waheed Y, Tahir S, Ahmad T and Qadri I:
Sequence comparison and phylogenetic analysis of core gene of
hepatitis C virus from Pakistani population. Afr J Biotechnol.
9:4561–4567. 2010.
|
|
13
|
Wu S, Skolnick J and Zhang Y: Ab initio
modeling of small proteins by iterative TASSER simulations. BMC
Biol. 5:172007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
de Vries SJ, van Dijk M and Bonvin AM: The
HADDOCK web server for data-driven biomolecular docking. Nat
Protoc. 5:883–897. 2010.PubMed/NCBI
|
|
15
|
Prevelige PE Jr: Inhibiting virus-capsid
assembly by altering the polymerisation pathway. Trends Biotechnol.
16:61–65. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Klein KC, Dellos SR and Lingappa JR:
Identification of residues in the hepatitis C virus core protein
that are critical for capsid assembly in a cell-free system. J
Virol. 79:6814–6826. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang X, Wagoner J, Negash A, Austin M,
McLauchlan J, Hahn YS, Rosen HR and Polyak SJ: Functional
characterization of core genes from patients with acute hepatitis
C. J Infect Dis. 201:912–922. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hourioux C, Ait-Goughoulte M, Patient R,
Fouquenet D, Arcanger-Doudet F, Brand D, Martin A and Roingeard P:
Core protein domains involved in hepatitis C virus-like particle
assembly and budding at the endoplasmic reticulum membrane. Cell
Microbiol. 9:1014–1027. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin W, Choe WH, Hiasa Y, Kamegaya Y,
Blackard JT, Schmidt EV and Chung RT: Hepatitis C virus expression
suppresses interferon signaling by degrading STAT1.
Gastroenterology. 128:1034–1041. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Akuta N, Suzuki F, Sezaki H, Suzuki Y,
Hosaka T, Someya T, Kobayashi M, Saitoh S, Watahiki S, Sato J,
Matsuda M, Kobayashi M, Arase Y, Ikeda K and Kumada H: Association
of amino acid substitution pattern in core protein of hepatitis C
virus genotype 1b high viral load and non-virological response to
interferon-ribavirin combination therapy. Intervirology.
48:372–380. 2005. View Article : Google Scholar
|
|
21
|
Akuta N, Suzuki F, Hirakawa M, Kawamura Y,
Yatsuji H, Sezaki H, Suzuki Y, Hosaka T, Kobayashi M, Kobayashi M,
Saitoh S, Arase Y, Ikeda K, Chayama K, Nakamura Y and Kumada H:
Amino acid substitution in hepatitis C virus core region and
genetic variation near the interleukin 28B gene predict viral
response to telaprevir with peginterferon and ribavirin.
Hepatology. 52:421–429. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hofmann WP, Polta A, Herrmann E, Mihm U,
Kronenberger B, Sonntag T, Lohmann V, Schönberger B, Zeuzem S and
Sarrazin C: Mutagenic effect of ribavirin on hepatitis C
nonstructural 5B quasispecies in vitro and during antiviral
therapy. Gastroenterology. 132:921–930. 2007. View Article : Google Scholar : PubMed/NCBI
|