Introduction
Metabolic syndrome (1) is a complex disorder, characterized by
a group of metabolic disorders, including hyperglycemia,
hypertension, hyperlipaemia and central obesity, which are the risk
factors of cardiovascular disease and diabetes. There is a high
prevalence of MS in developed countries. According to the national
health statistic reports issued in 2009 from the National Health
and Nutrition Examination Survey 2003–2006, the prevalence of
metabolic syndrome in the USA, is 34% in the population of 20 years
of age. The prevalence of metabolic syndrome increased in different
age groups for the two genders (2). However, in recent years, the
prevalence of MS has increased at an alarming rate in developing
countries and districts of Asia (3–5).
According to the National Health and Nutrition Examination Survey
2003–2006, Chinese prevalence of MS (4) increased by 14–18% in 2005 and has
continued to rise. Approximately 60–80% of diabetic patients are
also MS patients.
An unhealthy lifestyle, including lack of physical
exercise, bad nutritional habits, hormone changes and aging factors
are associated with the onset of MS (6). However the two important risk factors
for MS are central obesity and insulin resistance (7). Individuals with excess abdominal fat
are more likely to develop hypertension and increased levels of
blood cholesterol, triglycerides and fatty acids, which may affect
insulin in glucose regulation (7)
and lead to insulin resistance.
MS is caused by environmental and genetic factors.
Previous studies have revealed numerous target molecules and
variation associated with MS. Nuclear genes, including peroxisome
proliferator-activated receptor-γ, lamin A/C gene and IL-6, are
considered as susceptibility genes of MS (8–12).
Genetic factors, including mitochondrial DNA variants have also
been associated with the onset of MS (1,13,14).
Mitochondrial DNA (mtDNA) (15) contains 37 genes, including 2
ribosomal RNA genes, 22 transfer RNA genes and 13 coding genes,
which code the proteins involved in oxidative phosphorylation.
mtDNA has a higher mutation rate than nuclear DNA as it lacks
protective histones and is susceptible to oxidative damage from
reactive oxygen species (ROS) generated during respiration in the
mitochondria (16). Studies have
indicated that mitochondrial genetic defects may lead to β-cell
dysfunction and insulin resistance (17). Moreover, mitochondrial dysfunction
and biogenesis may be involved in the development of MS (18,19).
In the present study, a juvenile-onset metabolic
syndrome male with a maternally inherited diabetes family was
examined. In the patient’s family, a number of members diagnosed
with type 2 diabetes also presented characteristics of MS, however,
the 18-year-old male had not developed diabetes. Notably, a
heteroplasmic mtDNA mutation, A8890G, observed in the male was not
observed in other family members. The A8890G is located in the
ATPase 6 gene, which encodes for a subunit of ATP synthase (ATPase)
in mitochondria. Therefore, it is hypothesized that A8890G may be
associated with mitochondrial dysfunction and may contribute to
juvenile-onset of MS.
Materials and methods
Subjects
A maternally inherited diabetes family was contacted
through the Endocrinology Department and the Medical Examination
Center of the First Affiliated Hospital of Wenzhou Medical College
of Zhejiang Province in China and recruited. Peripheral blood
samples were obtained from the patient and all participating family
members. Serum triacylglycerol (TG), total cholesterol (TC),
low-density lipoprotein (LDL), high-density lipoprotein (HDL),
glucose, liver profile and kidney function parameters were
determined using routine automated assay methods following an
overnight fast.
Diagnosis of MS was determined according to the
diagnostic criteria proposed by the International Diabetes
Federation (IDF) in 2005 (6).
Subjects were diagnosed with metabolic syndrome if they had a waist
circumference of >90 cm in males and >80 cm in females and
two or more of the following four components: i) serum
triglycerides of >1.7 mmol/l or having clinical treatment; ii)
HDL-cholesterol levels <0.9 mmol/l in males and <1.0 mmol/l
in females or having clinical treatment; iii) BP of ≥130/85 mmHg or
under anti-hypertensive treatment; iv) fasting glucose of ≥5.6
mmol/l or known treatment for diabetes. Subjects with ≤2 risk
components were excluded.
A total of 165 freshmen were recruited in Wenzhou
Medical College and underwent a routine healthy check-up. The study
was approved by the Hospital Ethics Committee. Informed consent was
obtained from all participating subjects.
Mitochondrial DNA analysis
Genomic DNA was extracted from peripheral blood
leukocytes of each individual using universal genomic DNA
extraction kit version 3.0 (Takara Bio Inc., Shiga, Japan). Entire
mitochondrial DNA was amplified in 24 overlapping fragments using
primer sets, as described in a previous study (20).
PCR was performed at a final volume of 22 μl as
follows: 1.5 μl MgCl2 (25 mmol/l), 2.0 μl dNTP mixture
(2.5 mmol/l of each component), 2.0 μl 10X Ex Taq buffer, 0.2 μl of
each primer (10 μmol/l), 0.1 μl Ex Taq polymerase (Takara Bio Inc.)
and 2.0 μl genomic DNA as the template. PCR was performed in a
MyCycler Thermal Cycler 170–9703 (Bio-Rad, Hercules, CA, USA).
PCR conditions were as follows: initial denaturation
at 94ºC for 5 min, followed by 35 cycles of denaturation at 94ºC
for 30 sec, annealing at 60ºC for 45 sec and extension at 72ºC for
1 min, with a final extension at 72ºC for 6 min. Amplification
products were confirmed using electrophoresis in 1.2% agarose gels
and visualized by staining with ethidium bromide. PCR products
underwent direct sequencing in an ABI 3730 DNA sequencer (Sigma,
St. Louis, MO, USA). Sample sequences were compared with the
revised Cambridge Reference Sequence (rCRS) from Mitomap, a human
mitochondrial genome database (http://www.mitomap.org).
Multiple amino acid sequence alignment was performed
by Clustal X. The secondary structure of mitochondrial ATPase6 was
predicted by the SOSUI system (http://bp.nuap.nagoya-u.ac.jp/sosui/sosui_submit.html).
Denaturing high-performance liquid
chromatography (DHPLC) assay
PCR amplification of mtDNA mt 8702–8982 (281 bp) was
performed at a final volume of 20 μl as follows: 0.1 μl Pyrobest
DNA Polymerase (Takara Bio Inc.), 2.0 μl 10X Pyrobest buffer
(containing Mg2+), 3.0 μl dNTP mixture, 0.5 μl of each
primer (10 μmol/l, DHPLC grade), 2.0 μl genomic DNA as template.
The primer sequences used were: forward:
5′-CCATACACAACACTAAAGGACGAA-3′ and reverse:
5′-TTGAATGAGTAGGCTGATGGTTT-3′. PCR conditions were as follows: an
initial denaturation at 95ºC for 5 min, followed by 35 cycles of
denaturation at 95ºC for 10 sec, annealing at 60ºC for 20 sec and
extension at 72ºC for 45 sec, with a final extension at 72ºC for 6
min. PCR products were denatured at 95ºC for 1 min and cooled to
4ºC at a rate of 1ºC/min to allow for heteroduplex formation. The
DHPLC assay was performed in a WAVE® nucleic acid
fragment system (Transgenomic Inc., Omaha, NE, USA).
Results
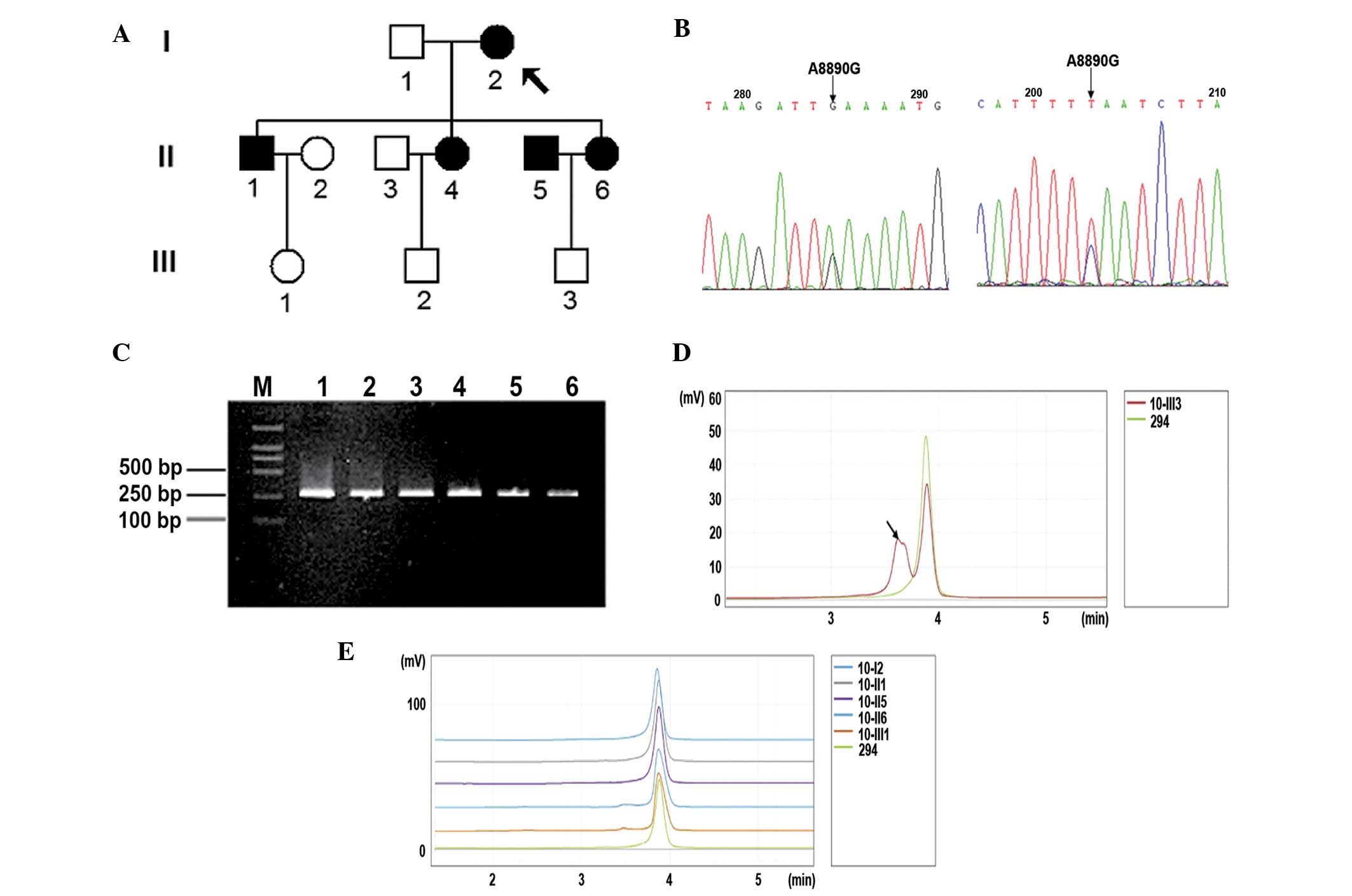
Family and biochemical analysis
Clinical and biochemical characteristics of all the
family members are provided in Table
I. The patient details presented are of a typical maternally
inherited diabetes family (Fig
1A).
 | Table IClinical and biochemical
characteristics of all family members. |
Table I
Clinical and biochemical
characteristics of all family members.
| Variables | Family members |
|---|
| General | I2 | II1 | II4 | II5 | II6 |
III1 |
III2 |
III3 |
| Gender | F | M | F | M | F | F | M | M |
| Age, years | 85 | 61 | 54 | 45 | 43 | 38 | 33 | 18 |
| Waist
circumstances, cm | 75 | 97 | 87 | 85 | 90 | 94 | 87 | 95 |
| BMI
(kg/m2) | 20.02 | 28.08 | 24.30 | 21.45 | 26.35 | 27.34 | 21.71 | 27.47 |
| DBP (mmHg) | 62 | 80 | 78 | 75 | 80 | 80 | 70 | 70 |
| SBP (mmHg) | 120 | 130 | 126 | 130 | 120 | 130 | 110 | 110 |
| History of
diabetes, years | 10 | 10 | 4 | 1 | 1 | 0 | 0 | 0 |
| Biochemical
parameters |
| FBG (mmol/l) | 4.8 | 7.78 | 5.17 | 6.98 | 10.78 | 5.87 | 5.08 | 5.08 |
| HbA1c1 (%) | 6.58 | 7.57 | 6.01 | 7.81 | 9.39 | 5.1 | 5.35 | 4.65 |
| BUN (mmol/l) | 5.7 | 4.1 | 6.9 | 4.3 | 3.9 | 6.1 | 4.3 | 4 |
| Cr (μmol/l) | 65 | 75 | 81 | 60 | 40 | 50 | 54 | 51 |
| UA (μmol/l) | 447 | 346 | 409 | 307 | 214 | 288 | 230 | 342 |
| Cys C (mg/l) | 1.41 | 1.32 | 1.57 | 1.28 | 0.68 | 1.20 | 1.09 | 1.29 |
| HB (mmol/l) | 0.07 | 0.06 | 0.07 | 0.05 | 0.12 | 0.09 | 0.06 | 0.07 |
| Lipid
parameters |
| TC (mmol/l) | 4.15 | 4.24 | 4.58 | 7.33 | 5.01 | 6.29 | 4.54 | 6.78 |
| TG (mmol/l) | 0.54 | 1.18 | 1.1 | 2.43 | 2.22 | 1.56 | 1.55 | 1.8 |
| HDL-C (mmol/l) | 1.4 | 1.3 | 1.36 | 1.24 | 0.96 | 1.59 | 1.04 | 0.97 |
| LDL-C (mmol/l) | 2.27 | 2.64 | 2.62 | 4.79 | 2.52 | 3.96 | 2.74 | 4.86 |
| apoA-I (g/l) | 1.2 | 1.38 | 1.41 | 1.07 | 1.24 | 1.53 | 0.94 | 1.13 |
| apoB-100 (g/l) | 0.98 | 1.15 | 1.13 | 1.62 | 1.11 | 1.29 | 0.93 | 2.14 |
|
apoA-I/apoB-100 | 1.22 | 1.2 | 1.25 | 0.66 | 1.12 | 1.19 | 1.01 | 0.53 |
| Hearing loss | + | + | + | | | | | |
| | | (Single ear) | − | − | − | − | − |
According to the IDF MS diagnostic criteria
(21,22), the 18-year-old male (III3) was
diagnosed with MS and other family members II1, II5, III6, III1 and
III3 also presented MS features. Subjects I2, II1, II4, II5 and II6
suffered from diabetes for >1 year, with hearing loss and mild
kidney impairment. Subjects II1, II6, III1 and III3 were overweight
(BMI 25–29 kg/cm2, Table
I). No obesity (BMI≥30) and hypertension was observed in the
family. In addition, it was observed that the male patient (III3)
and the patient’s father (II5) presented lipid metabolism
dysfunction with higher TG and LDL-C levels, lower HDL-C level and
<1.0 of the ApoA-I/ApoB100 ratio (Table I).
mtDNA variant analysis
Mitochondrial genomic DNA variants in the family are
provided in Table II. An
MS-associated variant T16189C was observed in this family. Using
sense-, antisense-sequencing and DHPLC analysis, a heteroplasmic
A-G substitution at mt 8890 (Fig. 1B
and C) was detected only in the 18-year-old male patient
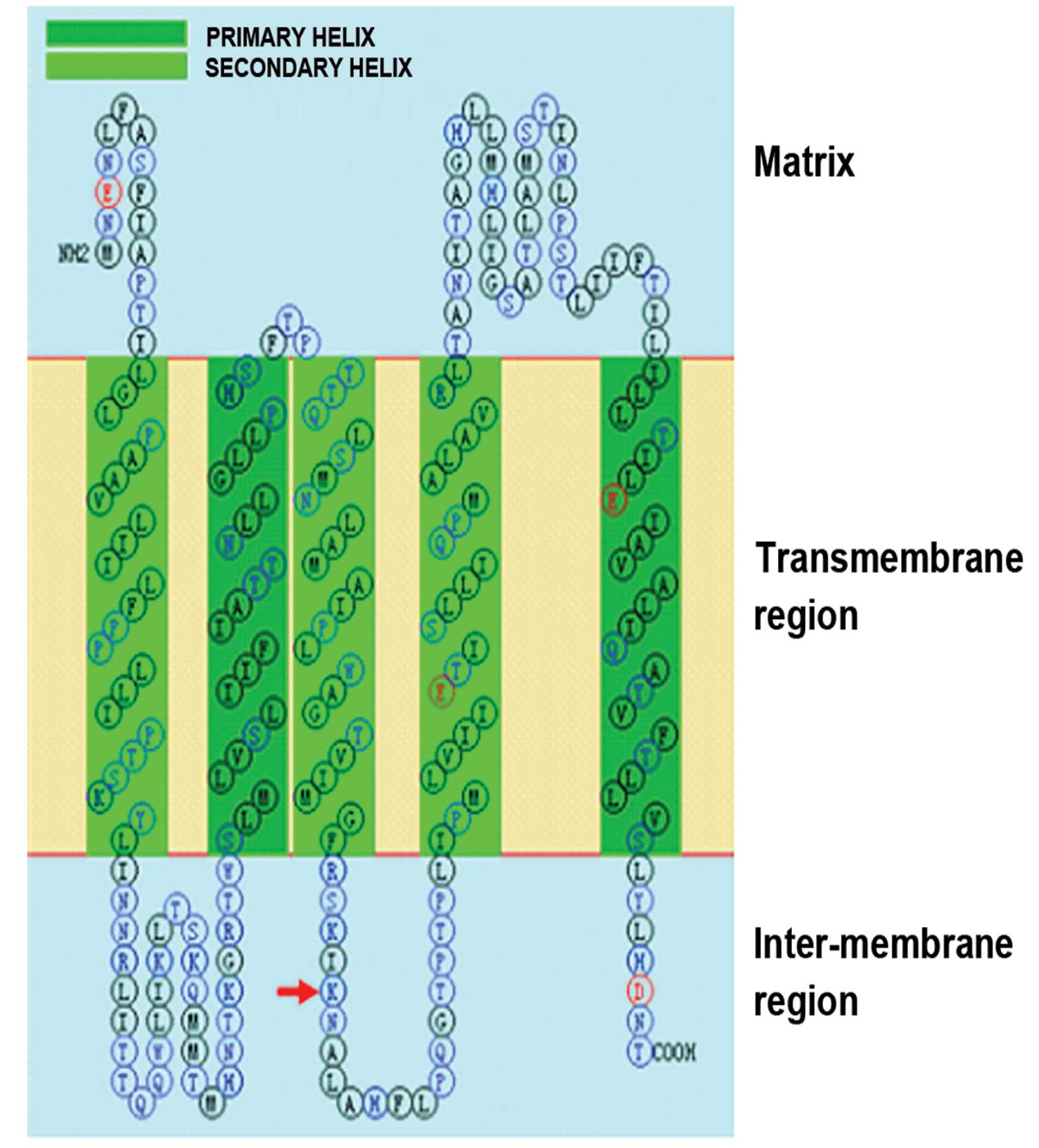
(III3). A8890G changed the basic amino acid Lys to Glu at position
122 of the ATPase6 subunit. This amino acid is located in the
intermembrane of the mitochondria (Fig. 2) and is highly conserved in 30
species (Table III). In
addition, there are numerous mtDNA variants, however A8890G was not
observed on the mitochondrial ATPase 6 gene of the control
subjects. No other MS or diabetes-associated mutations were
detected in this patient or the patient’s family.
| Table IImtDNA variants in this
family.a |
Table II
mtDNA variants in this
family.a
| Gene | Position | Nucleotide
replacement | Amino acid
replacement | Conservation
(H/B/M/X)b | Previously
Reportedc |
|---|
| D-Loop | 73 | A-G | | | Y |
| 263 | A-G | | | Y |
| 315 | C-CC | | | Y |
| 489 | T-C | | | Y |
| 16184 | C-T | | | Y |
| 16189 | T-C | | | Y |
| 16223 | C-T | | | Y |
| 16298 | T-C | | | Y |
| 16319 | G-A | | | Y |
| 12S rRNA | 750 | A-G | | A/A/A/- | Y |
| 1438 | A-G | | A/A/A/G | Y |
| 16S rRNA | 2706 | A-G | | A/G/A/A | Y |
| 2835 | C-T | | C/A/A/A | Y |
| ND2 | 4715 | A-G | | | Y |
| 4769 | A-G | | | Y |
| CO1 | 6179 | G-A | | | Y |
| 7028 | C-T | | | Y |
| 7196 | C-A | | | Y |
| CO2 | 8245 | A-G | | | Y |
| ATP6 | 8860 | A-G | Thr-Ala | T/A/A/T | Y |
| 8890 | A-G | Lys-Glu | K/K/K/N | N |
| 9053 | G-A | Ser-Asn | S/G/G/T | Y |
| CO3 | 9540 | T-C | | | Y |
| ND3 | 10398 | A-G | Thr-Ala | T/T/T/A | Y |
| 10400 | C-T | | | Y |
| ND4 | 10873 | T-C | | | Y |
| 11176 | G-A | | | Y |
| 11719 | G-A | | | Y |
| ND5 | 12705 | C-T | | | Y |
| ND6 | 14470 | T-C | | | Y |
| CytB | 15043 | G-A | | | Y |
| 15301 | G-A | | | Y |
| 15326 | A-G | Thr-Ala | T/M/I/I | Y |
| 15487 | A-T | | P/P/P/P | Y |
| Table IIIAmino acid in ATPase6 position 22 of
30 species. |
Table III
Amino acid in ATPase6 position 22 of
30 species.
| Species | Aminoacid |
|---|
| Gorilla
gorilla | Lys |
| Pan
paniscus | Lys |
| Pan
troglodytes | Lys |
| Pongo
pygmaeus | Lys |
| Hylobates
lar | Lys |
| Papio
hamadryas | Lys |
| Equus
caballus | Lys |
| Equus
asinus | Lys |
| Rhinoceros
unicornis | Lys |
| Ceratotherium
simum | Lys |
| Bos
taurus | Lys |
| Ovis
aries | Lys |
| Sus
scrofa | Lys |
| Balaenoptera
musculus | Lys |
| Balaenoptera
physalus | Lys |
| Hippopotamus
amphibius | Lys |
| Mus
musculus | Lys |
| Rattus
norvegicus | Lys |
| Myoxus
glis | Asn |
| Oryctolagus
cuniculus | Lys |
| Felis
catus | Lys |
| Halichoerus
grypus | Lys |
| Phoca
vitulina | Lys |
| Canis
familiaris | Lys |
| Artibeus
jamaicensis | Lys |
| Dasypus
novemcinctus | Lys |
| Didelphis
virginiana | Lys |
| Macropus
robustus | Lys |
| Ornithorhyncus
anatinus | Lys |
| Xenopus
laevis | Asn |
Discussion
Although nuclear genes are involved in MS onset,
mtDNA mutations are also significant in the development of MS.
Mitochondrial tRNAIle T4291C mutation was first observed
in a Caucasian population with MS where it was hypothesized to
cause a cluster of metabolic defects. In addition, mtDNA D-loop
T16189C was widely implicated in the development of insulin
resistance, MS and coronary artery disease (14,23).
Subsequent studies confirmed that mutations in mtDNA were
associated with diabetes, particularly, heteroplasmy
tRNALeu(UUR) A3243G was implicated in causing
maternal-inherited diabetes (24–26).
Extensive studies on the A3243G mutation revealed inefficient
aminoacylation, impairments in the processing of tRNA precursors
and base post-transcriptional modification of
tRNALeu(UUR), which induced mitochondrial dysfunction
(27–31).
In the present study, a juvenile-onset metabolic
syndrome male in a maternally inherited diabetes family was
examined. Although the family was a typical diabetic family,
characteristics of MS with mild increasing cystine C levels, a
sensitive marker for renal impairment, were also presented. The
patient presented central obesity (overweight and 95 cm of waist
circumference), dyslipidemia (high TG, low HDL level and low
apoA-I/apoB-100) and mild renal impairment according to cystine C
levels. Obesity is a risk factor for insulin resistance and
pancreatic β-cells are likely to secrete more insulin when
compensating for insulin resistance. The dysfunction of β-cells may
cause pre-diabetes, a condition ultimately leading to diabetes
(32).
Notably, the mtDNA genome assay detected a
heteroplasmic mtDNA mutation A8890G in the patient and was not
observed in other members of the family. In addition, MS-associated
mitochondrial T16189C single-nucleotide polymorphism was detected
in all the family members. No known diabetes or MS-associated
mitochondrial mutations, including tRNALeu(UUR)A3243G
and tRNAIleT4291C (1),
were detected.
A8890G is located in ATPase 6 gene, which encodes
for a subunit of ATP synthase in mitochondria. ATPase is also known
as mitochondrial respiratory chain complex V. It is a key enzyme in
cell energy conversion, participating in the synthesis and
hydrolysis of ATP. ATPase6 is one of the ATPase subunits and is
coded by the mitochondrial ATP6 gene. ATPase6 is a component of the
proton channel and is essential for proton transportation and
energy production (33,34). According to the mitomap database,
http://mitomap.org, A8890G has not been previously
reported. The majority of frequently reported mutations of ATPase 6
gene were heteroplasmic transversion T8993C and T8993G, which were
associated with Leigh and NARP syndrome. Symptom severity was
proportional to the heteroplasmy load (35–38).
Cell and mitochondrial function analysis indicated that mutations
led to energy deprivation, ROS overproduction and reduced ATP
production (39–41).
Unlike T8993C/G (Leu156Pro/Arg), which changed an
amino acid in the mitochondrial transmembrane protein, A8890G
changed the amino acid in the mitochondrial intermembrane protein,
which may affect the electrochemical proton gradient and energy
production. These changes may contribute to the development of MS.
Further evaluation of this abnormality is likely to include tissue
biopsy, mitochondrial function tests and cell analysis.
This mutation was not detected in other family
members, possibly as this mutation is a somatic mutation or a new
germline mutation. Mitochondrial replicative segregation during
cell division may affect the mutation (42). This indicates that the patient’s
mother (II6) may be a mosaic for this mutation. A low level of
A8890G mutant mitochondria that cannot be detected using present
analytical methods may be present in the patient’s mother. At
pre-ovulatory oocyte division, mutant and normal mitochondria
segregate into next generation oocytes at random, producing uneven
loading of mutant mitochondria. The patient may originate from an
oocyte with a higher heteroplasmy rate.
In conclusion, the novel heteroplasmic A8890G may
contribute to juvenile-onset MS. This mutation has not been
detected in other family members, possibly as it is a somatic
mutation or due to production of a new germline mutation or
mitochondrial genetic segregation in the juvenile-onset
patient.
Acknowledgements
Thanks for all the family members. This study is
supported by the National Key Technology R&D Program of China
(Grant no. 2009BAI80B02); the Science Foundation of Zhejiang
Province grants (nos. Y2090753, Y2100582); Zhejiang Provincial
Education Department Research grant (no. Y200804470).
Abbreviations:
|
MS
|
metabolic syndrome
|
|
ATPase6
|
ATP synthase subunit 6
|
|
mtDNA
|
mitochondrial DNA
|
|
DHPLC
|
denaturing high-performance liquid
chromatography
|
|
DBP
|
diastolic arterial blood pressure
|
|
SBP
|
systolic arterial blood pressure
|
|
FBG
|
fasting blood glucose
|
|
BUN
|
blood urine nitrogen
|
|
Cr
|
creatine
|
|
UA
|
urine acid
|
|
Cys C
|
cystine C
|
|
HB
|
β-hydroxybutyric acid
|
|
TC
|
total cholesterol
|
|
TG
|
triglycerol
|
|
HDL-C
|
high-density lipoprotein
cholesterol
|
|
LDL-C
|
low-density lipoprotein
cholesterol
|
References
|
1
|
Wilson FH, Hariri A, Farhi A, et al: A
cluster of metabolic defects caused by mutation in a mitochondrial
tRNA. Science. 306:1190–1194. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ervin RB: Prevalence of metabolic syndrome
among adults 20 years of age and over, by sex, age, race and
ethnicity and body mass index: United States, 2003–2006. Natl
Health Stat Report. 5:1–7. 2009.PubMed/NCBI
|
|
3
|
Lim S, Shin H, Song JH, et al: Increasing
prevalence of metabolic syndrome in Korea: the Korean National
Health and Nutrition Examination Survey for 1998–2007. Diabetes
Care. 34:1323–1328. 2011.
|
|
4
|
Gu D, Reynolds K, Wu X, et al: Prevalence
of the metabolic syndrome and overweight among adults in China.
Lancet. 365:1398–1405. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yi Z, Jing J, Liu X-Y, et al: Prevalence
of the metabolic syndrome among rural original adults in NingXia,
China. BMC Public Health. 10:1402010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alberti KG, Zimmet P and Shaw J; IDF
Epidemiology Task Force Consensus Group. The metabolic syndrome - a
new worldwide definition. Lancet. 366:1059–1062. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Janssen I, Katzmarzyk PT and Ross R: Waist
circumference and not body mass index explains obesity-related
health risk. Am J Clin Nutr. 79:379–384. 2004.PubMed/NCBI
|
|
8
|
Ordovas JM: Nutrigenetics, plasma lipids
and cardiovascular risk. J Am Diet Assoc. 106:1074–1081; quiz 1083.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Frederiksen L, Brødbaek K, Fenger M, et
al: Comment: studies of the Pro12Ala polymorphism of the PPAR-gamma
gene in the Danish MONICA cohort: homozygosity of the Ala allele
confers a decreased risk of the insulin resistance syndrome. J Clin
Endocrinol Metab. 87:3989–3992. 2002.PubMed/NCBI
|
|
10
|
Bennet AM, Prince JA, Fei GZ, et al:
Interleukin-6 serum levels and genotypes influence the risk for
myocardial infarction. Atherosclerosis. 171:359–367. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murase Y, Yagi K, Katsuda Y, et al: An
LMNA variant is associated with dyslipidemia and insulin resistance
in the Japanese. Metabolism. 51:1017–1021. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Steinle NI, Kazlauskaite R, Imumorin IG,
et al: Variation in the lamin A/C gene: associations with metabolic
syndrome. Arterioscler Thromb Vasc Biol. 24:1708–1713. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Juo SH, Lu MY, Bai RK, et al: A common
mitochondrial polymorphism 10398A>G is associated metabolic
syndrome in a Chinese population. Mitochondrion. 10:294–299. 2010.
View Article : Google Scholar
|
|
14
|
Palmieri VO, De Rasmo D, Signorile A, et
al: T16189C mitochondrial DNA variant is associated with metabolic
syndrome in Caucasian subjects. Nutrition. 27:773–777. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Anderson S, Bankier AT, Barrell BG, et al:
Sequence and organization of the human mitochondrial genome.
Nature. 290:457–465. 1981. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yakes FM and Van Houten B: Mitochondrial
DNA damage is more extensive and persists longer than nuclear DNA
damage in human cells following oxidative stress. Proc Natl Acad
Sci USA. 94:514–519. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Soejima A, Inoue K, Takai D, et al:
Mitochondrial DNA is required for regulation of glucose-stimulated
insulin secretion in a mouse pancreatic beta cell line, MIN6. J
Biol Chem. 271:26194–26199. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishio Y, Kanazawa A, Nagai Y, et al:
Regulation and role of the mitochondrial transcription factor in
the diabetic rat heart. Ann NY Acad Sci. 1011:78–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nisoli E, Clementi E, Carruba MO, et al:
Defective mitochondrial biogenesis: a hallmark of the high
cardiovascular risk in the metabolic syndrome? Circ Res.
100:795–806. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rieder MJ, Taylor SL, Tobe VO, et al:
Automating the identification of DNA variations using quality-based
fluorescence re-sequencing: analysis of the human mitochondrial
genome. Nucleic Acids Res. 26:967–973. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zimmet P, Alberti KG, Kaufman F, et al:
The metabolic syndrome in children and adolescents. Lancet.
370:1541–1542. 2007.
|
|
22
|
Zimmet P, Alberti KG, Kaufman F, et al:
The metabolic syndrome in children and adolescents-an IDF consensus
report. Pediatr Diabetes. 8:299–306. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mueller EE, Eder W, Ebner S, et al: The
mitochondrial T16189C polymorphism is associated with coronary
artery disease in Middle European populations. PLoS One.
6:e164552011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van den Ouweland JM, Lemkes HH, Ruitenbeek
W, et al: Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large
pedigree with maternally transmitted type II diabetes mellitus and
deafness. Nat Genet. 1:368–371. 1992.PubMed/NCBI
|
|
25
|
Ohkubo K, Yamano A, Nagashima M, et al:
Mitochondrial gene mutations in the tRNA(Leu(UUR)) region and
diabetes: prevalence and clinical phenotypes in Japan. Clin Chem.
47:1641–1648. 2001.PubMed/NCBI
|
|
26
|
Maassen JA, ‘T Hart LM, Van Essen E, et
al: Mitochondrial diabetes: molecular mechanisms and clinical
presentation. Diabetes. 53(Suppl 1): S103–S109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rossmanith W and Karwan RM: Impairment of
tRNA processing by point mutations in mitochondrial tRNA(Leu)(UUR)
associated with mitochondrial diseases. FEBS Lett. 433:269–274.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Janssen GM, Maassen JA and van Den
Ouweland JM: The diabetes-associated 3243 mutation in the
mitochondrial tRNA(Leu(UUR) gene causes severe mitochondrial
dysfunction without a strong decrease in protein synthesis rate. J
Biol Chem. 274:29744–29748. 1999. View Article : Google Scholar
|
|
29
|
Chomyn A, Enriquez JA, Micol V, et al: The
mitochondrial myopathy, encephalopathy, lactic acidosis and
stroke-like episode syndrome-associated human mitochondrial
tRNALeu(UUR) mutation causes aminoacylation deficiency and
concomitant reduced association of mRNA with ribosomes. J Biol
Chem. 275:19198–19209. 2000. View Article : Google Scholar
|
|
30
|
Yasukawa T, Suzuki T, Ueda T, et al:
Modification defect at anticodon wobble nucleotide of mitochondrial
tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial
myopathy, encephalopathy, lactic acidosis and stroke-like episodes.
J Biol Chem. 275:4251–4257. 2000. View Article : Google Scholar
|
|
31
|
Park H, Davidson E and King MP: The
pathogenic A3243G mutation in human mitochondrial tRNALeu(UUR)
decreases the efficiency of aminoacylation. Biochemistry.
42:958–964. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee JM: Why young adults hold the key to
assessing the obesity epidemic in children. Arch Pediatr Adolesc
Med. 162:682–687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hutcheon ML, Duncan TM, Ngai H, et al:
Energy-driven subunit rotation at the interface between subunit a
and the c oligomer in the F(O) sector of Escherichia coli
ATP synthase. Proc Natl Acad Sci USA. 98:8519–8524. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stock D, Leslie AG and Walker JE:
Molecular architecture of the rotary motor in ATP synthase.
Science. 286:1700–1705. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baracca A, Sgarbi G, Mattiazzi M, et al:
Biochemical phenotypes associated with the mitochondrial ATP6 gene
mutations at nt8993. Biochim Biophys Acta. 1767:913–919. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Santorelli FM, Tanji K, Shanske S, et al:
Heterogeneous clinical presentation of the mtDNA NARP/T8993G
mutation. Neurology. 49:270–273. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Takahashi S, Makita Y, Oki J, et al: De
novo mtDNA nt 8993 (T→G) mutation resulting in Leigh syndrome. Am J
Hum Genet. 62:717–719. 1998.PubMed/NCBI
|
|
38
|
Tatuch Y, Christodoulou J, Feigenbaum A,
et al: Heteroplasmic mtDNA mutation (T→G) at 8993 can cause Leigh
disease when the percentage of abnormal mtDNA is high. Am J Hum
Genet. 50:852–858. 1992.
|
|
39
|
Mattiazzi M, Vijayvergiya C, Gajewski CD,
et al: The mtDNA T8993G (NARP) mutation results in an impairment of
oxidative phosphorylation that can be improved by antioxidants. Hum
Mol Genet. 13:869–879. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sgarbi G, Baracca A, Lenaz G, et al:
Inefficient coupling between proton transport and ATP synthesis may
be the pathogenic mechanism for NARP and Leigh syndrome resulting
from the T8993G mutation in mtDNA. Biochem J. 395:493–500. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tatuch Y and Robinson BH: The
mitochondrial DNA mutation at 8993 associated with NARP slows the
rate of ATP synthesis in isolated lymphoblast mitochondria. Biochem
Biophys Res Commun. 192:124–128. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shoubridge EA: Mitochondrial DNA
segregation in the developing embryo. Hum Reprod. 15(Suppl 2):
S229–S234. 2000. View Article : Google Scholar
|