Introduction
Cardiac hypertrophy frequently progresses into
chronic heart failure (CHF). Slowing or reversing cardiac
remodeling is an important therapeutic goal in patients with CHF.
Studies have shown that hydroxymethylglutaryl-CoA reductase
inhibitors (statins) attenuate cardiac remodeling in animals or
patients with either ischemic or non-ischemic CHF (1–7),
suggesting that statin therapy may be a potential novel approach
for CHF. A meta-analysis of randomized controlled trials showed
that treatment with statins in CHF patients attenuates cardiac
remodeling and relives clinical symptoms (8).
Pleiotropic effects of statins have been extensively
investigated; however, the involvement of statins in extrinsic and
intrinsic apoptosis pathways during CHF remains largely unknown. We
previously demonstrated that pressure overload induced prolonged
endoplasmic reticulum (ER) stress, which contributes to
cardiomyocyte apoptosis during the progression of cardiac
hypertrophy to CHF (9). In
addition, we demonstrated that the inhibition of cardiac remodeling
by statins is associated with amelioration of ER stress-initiated
apoptosis via decreasing the expression of C/EBP homologous protein
(CHOP) (1). When ER stress is
prolonged, however, initiation of the apoptotic processes is
promoted by CHOP and also by the activation of c-Jun N-terminal
kinases (JNK)- and/or caspase-12-dependent pathways (10). The intrinsic apoptotic signaling
pathway may be initiated by mitochondrial events and/or the ER
(11,12). Pro-apoptotic proteins, caspase-12
and Bax, are closely associated with mitochondrial events and ER
stress during apoptosis. Bax is phosphorylated by stress-activated
c-Jun N-terminal kinase (JNK), which leads to mitochondrial
translocation prior to apoptosis (13). The extrinsic apoptosis pathway
initiated by tumor necrosis factor α (TNF-α) is critical in CHF and
the receptor (extrinsic) and the mitochondrial (intrinsic) pathway
are interconnected at different levels. However, it remains unknown
whether or not statins inhibit cardiac remodeling through
interfering with the TNF-α-JNK related signaling pathway.
Therefore, it was hypothesized that pravastatin
delayed the progression of cardiac hypertension to CHF by
inhibiting the JNK-mediated apoptotic signal pathway. To confirm
this hypothesis, the involvement of pravastatin on the intrinsic
pro-apoptotic proteins, caspase-12 and Bax, in vivo and
in vitro was investigated. Furthermore, as JNK is important
in the intrinsic apoptotic signaling pathway, the effect of
pravastatin on JNK in mouse hearts subjected to TAC and cultured
cardiomyocytes stimulated by TNF-α were analyzed.
Materials and methods
Animal models and experimental
protocols
All procedures were conducted in male C57BL/6J mice
(age, 7–8 weeks; weight, 20–24 g, provided by the Animal Center of
Southern Medical University), were approved by the Animal Care and
Use Committee of the Southern Medical University (Guangzhou,
Guangdong, China) and were in accordance with the Guide for the
Care and Use of Laboratory Animals published by the US National
Institutes of Health (NIH publication no. 85–23, revised 1996).
C57BL/6J mice were anesthetized with a combination
of ketamine (100 mg/kg) and xylazine (5 mg/kg) via intraperitoneal
injection. Transverse aortic constriction (TAC) surgery was
performed as previously described (14,15).
Pravastatin (Pra, 5 or 20 mg/kg, dissolved in 0.9% saline; provided
by Daiichi-Sankyou Pharmaceutical Co. Ltd., Tokyo, Japan) was
orally administered from the third day post-surgery. The mice were
divided into four groups: Sham-operated (n=7), TAC (n=8), TAC +
Pra5 (n=5) and TAC + Pra20 (n=6) groups. Mice were sacrificed by
anesthesia overdose with 150 mg/kg pentobarbital sodium
intraperitoneal and cervical dislocation following analysis of
cardiac functions and hypertrophy with echocardiography and
invasive left ventricular (LV) hemodynamic assessment 4 weeks after
TAC. The hearts and lungs were harvested and weighed. The hearts
and lungs used for western blot analysis were snap-frozen in liquid
nitrogen and stored at −80°C.
Echocardiography
Transthoracic echocardiography was performed with a
Sonos 4500 and a 15–6 L MHz transducer (Philips, Eindhoven, The
Netherlands). Images were standardized to short axis view at the LV
mid-papillary level and the posterior wall diastolic thickness
(LVPWd), LV end-diastolic diameter (LVEDd) and LV end-systolic
diameter (LVESd) were recorded. LV systolic function was also
assessed from these measurements by calculating the LV fractional
shortening (LVFS) and the LV ejection fraction (LVEF).
Hemodynamic measurement
Pressure overload was confirmed by measuring the
gradient in carotid artery pressure by invasive hemodynamic
assessment; the systolic blood pressure gradient was identified to
be similar between TAC and TAC with pravastatin groups. For LV
hemodynamic assessment, mice were anesthetized, intubated and
ventilated. A Millar catheter (Millar Instruments, Inc., Houston,
TX, USA) was inserted into the right carotid artery and advanced
into the LV cavity. LV systolic pressure (LVSP) and the LV
end-diastolic pressure (LVEDp) were recorded. The maximum and
minimum rates of LV pressure change (dP/dt max and dP/dt min,
respectively), as well as contractility index (max dp/dt divided by
corresponding LV pressure) and diastolic index (min dp/dt divided
by corresponding LV pressure) were calculated using using PowerLab
software (blood pressure module, ADInstruments Shanghai Trading Co,
Shanghai, China).
Neonatal cardiomyocyte culture
Ventricular myocytes were prepared from
Sprague-Dawley rats (age, 1–2 days) obtained from the Animal center
of Southern Medical University. In brief, the rats were sacrificed
by 2% isoflurane inhalation and cervical dislocation. The hearts
were quickly excised and immediately embedded in freezing Hank’s
solution. Cardiomyocytes were dispersed by digestion with 0.1%
trypsin and 0.03% collagenase at 37°C, then were collected after
differential adhesion of non-cardiomyocytes and plated at a density
of 150–200 cells/mm2. Cardiomyocytes were incubated for
72 h in Dulbecco’s modified Eagle’s medium supplemented with 10%
fetal calf serum and then grown for 24 h under serum-free
conditions. Pravastatin (10 μM) was added 2 h prior to the addition
of 10 ng/ml TNF-α.
Western blot analysis
Protein samples were prepared from whole-heart
homogenates or cultured cardiomyocytes. A total quantity of 30 μg
of each sample was separated on 5–15% gradient polyacrylamide gels.
When transferred onto nitrocellulose membranes, the membranes were
incubated with primary antibodies against caspase-12, JNK,
phospho-JNK, Bax and glyceraldehyde 3-phosphate dehydrogenase
(GAPDH; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA)
followed by incubation with horseradish peroxidase-conjugated
secondary antibodies (Santa Cruz Biotechnology, Inc.). The bound
antibody was detected by the enhanced chemiluminescence method
according to the manufacturer’s instructions (Amersham Bioscience,
Buckinghamshire, UK) and band intensities were quantified by the
use of NIH image software (Image J 1.42q; NIH, Bethesda, MD,
USA).
Statistical analysis
All data are presented as the mean ± SE. P<0.05
was considered to indicate a statistically significant difference.
Unpaired Student’s t-test was used for comparisons between two
groups and one-way analysis of variance with post hoc analysis
using the Tukey-Kramer test was used for multiple comparisons.
Results
Pravastatin attenuates cardiac
remodeling
It was demonstrated that pravastatin improved
cardiac remodeling and dysfunction induced by TAC (Fig. 1A). Prior to TAC, the LV wall
thickness and dimensions were similar in the four groups of mice
(data not shown). TAC induced significant LV hypertrophy and
dilation (Fig. 1B and C). Four
weeks following TAC, the heart became larger and the heart
weight/body weight ratio increased significantly compared with the
sham-operated group (Fig. 1D and
E). LV wall thickness (Fig.
1B) and dimensions (Fig. 1C),
measured by echocardiography, were greater in the TAC group
compared with the sham-operated group. Pravastatin treatment
significantly inhibited LV hypertrophy and dilation in a
dose-dependent manner (Fig.
1A–E).
 | Figure 1Effects of pravastatin on cardiac
remodeling 4 weeks following surgery. (A) Representative
echocardiograms of the LV in sham, TAC and TAC + pravastatin 5 and
20 mg/kg/d groups (Pra5 and Pra20, resepctively). (B) Results of
LVPWd. (C) LV dimensions (LVEDd and LVESd). (D) HW/BW ratio. (E)
Representative images of the heart from different groups. Scale
bar, 2 cm. Arrows indicate banding site of aortic arch. n=8 in sham
and TAC groups, n=6 in TAC + Pra5 and Pra20 groups, respectively
for Fig. 1B and C; n=7, 8, 6 and 6 in the respective groups for
Fig. 1D. *P<0.01 and #P<0.05 vs. the
TAC group. LV, left ventricle; TAC, transverse aortic constriction;
LVPWd, left ventricular posterior wall diastolic thickness; LVEDd,
left ventricular end-diastolic diameter; LVESd, left ventricular
end-systolic diameter; HW, heart weight; BW, body weight. |
Pravastatin improves heart failure
Heart function was analyzed by echocardiography, LV
hemodynamics and pulmonary congestion. The hemodynamic measurements
obtained 4 weeks after TAC, confirmed that LV pressure was
significantly increased and LV function indexes were decreased, as
systolic function evaluated by dP/dt max, ratio of dP/dt max to
instantaneous pressure and diastolic function by dP/dt min, ratio
of dP/dt min to instantaneous pressure (Fig. 2A–C). Pravastatin treatment did not
significantly decrease LV pressure or increase LV systolic
function, but improved LV diastolic function, as reflected by LVEDP
and diastolic index (P<0.05; Fig.
2A and B). FS and EF, parameters of LV systolic function,
decreased significantly in the TAC group compared with the sham
group. The lung weight/body weight (LW/BW) ratio, an index of
pulmonary congestion, was markedly higher in TAC mice than in
sham-operated mice, while pravastatin-treated TAC mice exhibited a
significantly higher FS and EF as well as a lower LW/BW ratio
(P<0.05 or P<0.01; Fig. 2C and
D).
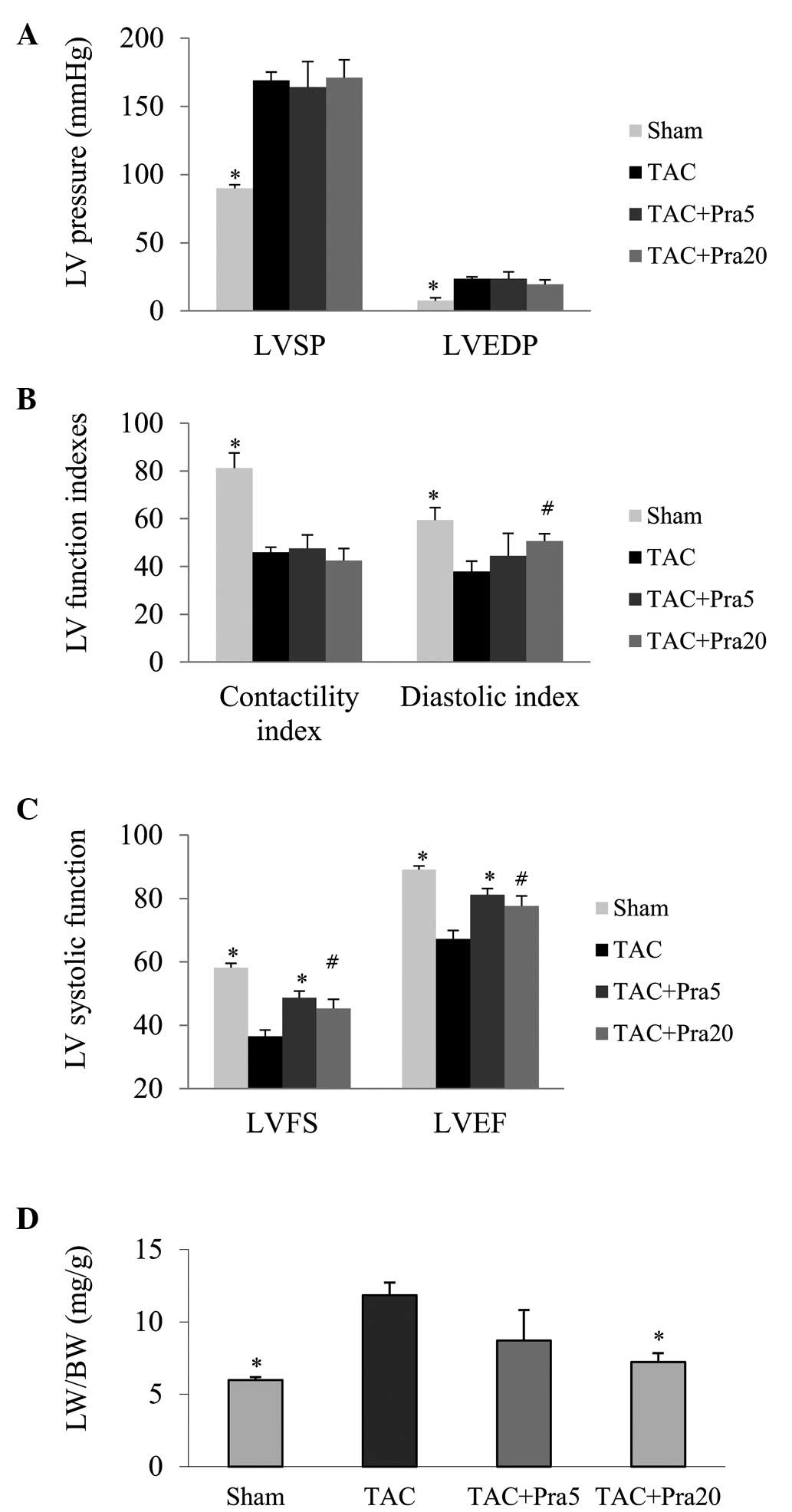 | Figure 2Effects of pravastatin on cardiac
function 4 weeks following surgery. (A) Left ventricular peak
systolic and end-diastolic pressure measured by using a Millar
catheter. (B) LV contractility and diastolic indices. (C) LV
systolic function (LVFS and LVEF) analyzed by echocardiography. (D)
Pulmonary congestion determined by the LW/BW ratio.
*P<0.01 and #P<0.05 vs. the TAC group;
n=7, 8, 5 and 6 in the sham, TAC, TAC + Pra5 and TAC + Pra20
groups, respectively for Fig. 2A and B; n=8, 8, 6 and 6 in the
respective groups for Fig. 2C; and n=7, 8, 6 and 6 in the
respective group for Fig. 2D. LVSP, left ventricular peak systolic
pressure; LVEDP, left ventricular end-diastolic pressure; LVFS, LV
fractional shortening; LVEF, LV ejection fraction; LW/BW, lung
weight/body weight; TAC, transverse aortic constriction. |
Pravastatin inhibits JNK phosphorylation
in vivo
As JNK is important in the intrinsic pro-apoptotic
signaling pathway, it was investigated whether pravastatin
influences the JNK signaling pathway in TAC mice. It was
demonstrated that the ratio of phosphorylated-JNK to JNK was
significantly greater in the TAC mice than in the sham-operated
group, while treatment with pravastatin markedly decreased the
activity of JNK (Fig. 3A and
B).
Pravastatin reduces the protein
expression levels of caspase-12 and Bax in vivo
As shown in Fig. 4,
the protein expression of caspase-12 and Bax was significantly
higher four weeks following TAC compared with the sham-operated
group; while pravastatin treated TAC mice exhibited significantly
lower expression levels of these apoptosis-related proteins.
Pravastatin inhibits the apoptosis
signaling pathway in cultured cardiomyocytes
In cultured neonatal rat cardiomyocytes, treatment
with TNF-α for 24 h significantly activated intrinsic apoptotic
signaling, as determined by the increase of caspase-12 and Bax
proteins, and the activity of JNK. Co-treatment with 10 μM
pravastatin markedly decreased the expression of these
apoptosis-related proteins and also downregulated the expression of
phosphorylated-JNK (Fig. 5A and
B). Thus, TNF-α is involved in cardiac molecular and cellular
changes during TAC and is associated with heart failure.
Discussion
This study demonstrated that pravastatin exerts
cardioprotection against cardiac remodeling by the inhibition of
the JNK-dependent intrinsic pro-apoptotic signaling pathway in TAC
mice, which supports the hypothesis that inhibition of JNK
phosphorylation is a potential therapeutic target for slowing the
progression of hypertrophy to heart failure.
JNK, a predominant branch of the mitogen-activated
protein kinase signaling cascades, has been implicated in the
pathophysiology of cardiac hypertrophy and heart failure (16,17).
Myocardial JNK1/2 is activated by inflammatory cytokines, oxidant
stress, G protein-coupled receptors and ER stress (18–24),
and all of which are associated with myocardial hypertrophy and
heart failure. To the best of our knowledge, the involvement of JNK
in cardiac remodeling and cardiomyocyte apoptosis remains
controversial (17). Previous
studies have clearly established the involvement of JNK in
TNF-α-induced apoptosis, stimulating the release of cytochrome
c from mitochondria through an analogous pathway involving
the proapoptotic proteins Bid and Bax. Moreover, JNK may be
activated prior to or following the induction of ER stress and then
induces the activation of caspase-12, which is central in ER
stress-induced apoptosis (25–28).
The activation of JNK and caspase-12 is associated with the TNF
receptor associated factor-2, while TNF-α is an important
therapeutic target of statins (29). However, it remains to be determined
whether statins improve cardiac remodeling thorough the modulation
of TNF-α associated apoptosis initiated at the ER and
mitochondria.
In the present study, pravastatin was observed to
significantly inhibit the activation of JNK and the upregulation of
pro-apoptotic proteins Bax and cleavage caspase-12 in TAC hearts
and TNF-α stimulated cardiomyocytes. JNK has been shown to be
critical for the release of cytochrome c from mitochondria
(30), and may also be essential
for ER stress-induced cardiomyocyte apoptosis. Bax and caspase-12
may be stimulated by JNK in cardiomyocytes. The activated Bax forms
membrane channels through which cytochrome c is released
(31), while caspase-12 was
demonstrated to be critical in response to ER stress-induced
apoptosis (25).
An increasing number of studies suggest that
apoptosis may be a key modulator in the transition from
compensatory hypertrophy to heart failure. Mitochondrial
dysfunction and ER stress are demonstrated to be involved in the
intrinsic apoptosis signaling pathway. Caspase-12 and Bax are two
important proteins, which indicate ER stress and mitochondrial
events respectively during cardiomyocyte apoptosis. The results of
the present study demonstrated that caspase-12 and Bax in TAC mice
were inhibited by pravastatin. Also, in cultured cardiomyocytes,
pravastatin was shown to inhibit TNF-α-induced caspase-12 and Bax,
which may contribute to cardiomyocyte hypertrophy. TNF-α, a
proinflammatory cytokine, induces cardiomyocyte hypertrophy,
migration, apoptosis and necrosis, which results in ventricular
remodeling and heart failure (32). The results demonstrated that
pravastatin inhibits cardiac remodeling and improves cardiac
function via regulating TNF-α associated JNK-dependent intrinsic
apoptosis signaling and thus slows the progression of hypertrophy
to heart failure. This may suggest the use of JNK-targeted drugs as
an alternative therapeutic strategy for patients with cardiac
hypertrophy and heart failure.
In conclusion, the results of this study indicate
that pravastatin attenuates cardiac remodeling by inhibiting
JNK-dependent pro-apoptotic signaling.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81170146, to Y.L.),
the Team Program of Natural Science Foundation of Guangdong
Province, China (grant no. S2011030003134, to Y.L. and J.B.).
Abbreviations:
|
CHF
|
chronic heart failure
|
|
CHOP
|
C/EBP homologous protein
|
|
ER
|
endoplasmic reticulum
|
|
TAC
|
transverse aortic constriction
|
|
TNF-α
|
tumor necrosis factor-α
|
|
JNK
|
c-Jun N-terminal kinase
|
|
LVPWd
|
left ventricular posterior wall
diastolic thickness
|
|
LVEDd
|
LV end-diastolic diameter
|
|
LVESd
|
LV end-systolic diameter
|
|
LVFS
|
LV fractional shortening
|
|
LVEF
|
LV ejection fraction
|
References
|
1
|
Zhao H, Liao Y, Minamino T, et al:
Inhibition of cardiac remodeling by pravastatin is associated with
amelioration of endoplasmic reticulum stress. Hypertens Res.
31:1977–1987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Horwich TB, MacLellan WR and Fonarow GC:
Statin therapy is associated with improved survival in ischemic and
non-ischemic heart failure. J Am Coll Cardiol. 43:642–648. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fukuta H, Sane DC, Brucks S and Little WC:
Statin therapy may be associated with lower mortality in patients
with diastolic heart failure: a preliminary report. Circulation.
112:357–363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sola S, Mir MQ, Lerakis S, Tandon N and
Khan BV: Atorvastatin improves left ventricular systolic function
and serum markers of inflammation in nonischemic heart failure. J
Am Coll Cardiol. 47:332–337. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liao Y, Zhao H, Ogai A, et al:
Atorvastatin slows the progression of cardiac remodeling in mice
with pressure overload and inhibits epidermal growth factor
receptor activation. Hypertens Res. 31:335–344. 2008. View Article : Google Scholar
|
|
6
|
Takemoto M, Node K, Nakagami H, et al:
Statins as antioxidant therapy for preventing cardiac myocyte
hypertrophy. J Clin Invest. 108:1429–1437. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martin J: Statins and congestive heart
failure. Curr Atheroscler Rep. 10:369–376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang L, Zhang S, Jiang H, Sun A, Zou Y
and Ge J: Effects of statin treatment on cardiac function in
patients with chronic heart failure: a meta-analysis of randomized
controlled trials. Clin Cardiol. 34:117–123. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okada K, Minamino T, Tsukamoto Y, et al:
Prolonged endoplasmic reticulum stress in hypertrophic and failing
heart after aortic constriction: possible contribution of
endoplasmic reticulum stress to cardiac myocyte apoptosis.
Circulation. 110:705–712. 2004. View Article : Google Scholar
|
|
10
|
Oyadomari S, Araki E and Mori M:
Endoplasmic reticulum stress-mediated apoptosis in pancreatic
beta-cells. Apoptosis. 7:335–345. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xin W, Li X, Lu X, Niu K and Cai J:
Involvement of endoplasmic reticulum stress-associated apoptosis in
a heart failure model induced by chronic myocardial ischemia. Int J
Mol Med. 27:503–509. 2011.
|
|
13
|
Kim BJ, Ryu SW and Song BJ: JNK- and p38
kinase-mediated phosphorylation of Bax leads to its activation and
mitochondrial translocation and to apoptosis of human hepatoma
HepG2 cells. J Biol Chem. 281:21256–21265. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liao Y, Asakura M, Takashima S, et al:
Benidipine, a long-acting calcium channel blocker, inhibits cardiac
remodeling in pressure-overloaded mice. Cardiovasc Res. 65:879–888.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liao Y, Takashima S, Asano Y, et al:
Activation of adenosine A1 receptor attenuates cardiac hypertrophy
and prevents heart failure in murine left ventricular
pressure-overload model. Circ Res. 93:759–766. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ogut O and Brozovich FV: The potential
role of MLC phosphatase and MAPK signalling in the pathogenesis of
vascular dysfunction in heart failure. J Cell Mol Med.
12:2158–2164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rose BA, Force T and Wang Y:
Mitogen-activated protein kinase signaling in the heart: angels
versus demons in a heart-breaking tale. Physiol Rev. 90:1507–1546.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yano M, Kim S, Izumi Y, Yamanaka S and
Iwao H: Differential activation of cardiac c-jun amino-terminal
kinase and extracellular signal-regulated kinase in angiotensin
II-mediated hypertension. Circ Res. 83:752–760. 1998. View Article : Google Scholar
|
|
19
|
Seko Y, Takahashi N, Tobe K, Kadowaki T
and Yazaki Y: Pulsatile stretch activates mitogen-activated protein
kinase (MAPK) family members and focal adhesion kinase (p125(FAK))
in cultured rat cardiac myocytes. Biochem Biophys Res Commun.
259:8–14. 1999. View Article : Google Scholar
|
|
20
|
Ramirez MT, Sah VP, Zhao XL, Hunter JJ,
Chien KR and Brown JH: The MEKK-JNK pathway is stimulated by
alpha1-adrenergic receptor and ras activation and is associated
with in vitro and in vivo cardiac hypertrophy. J Biol Chem.
272:14057–14061. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choukroun G, Hajjar R, Fry S, et al:
Regulation of cardiac hypertrophy in vivo by the stress-activated
protein kinases/c-Jun NH(2)-terminal kinases. J Clin Invest.
104:391–398. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Esposito G, Prasad SV, Rapacciuolo A, Mao
L, Koch WJ and Rockman HA: Cardiac overexpression of a G(q)
inhibitor blocks induction of extracellular signal-regulated kinase
and c-Jun NH(2)-terminal kinase activity in in vivo pressure
overload. Circulation. 103:1453–1458. 2001. View Article : Google Scholar
|
|
23
|
Honsho S, Nishikawa S, Amano K, et al:
Pressure-mediated hypertrophy and mechanical stretch induces IL-1
release and subsequent IGF-1 generation to maintain compensative
hypertrophy by affecting Akt and JNK pathways. Circ Res.
105:1149–1158. 2009. View Article : Google Scholar
|
|
24
|
Bartha E, Solti I, Kereskai L, et al: PARP
inhibition delays transition of hypertensive cardiopathy to heart
failure in spontaneously hypertensive rats. Cardiovasc Res.
83:501–510. 2009. View Article : Google Scholar
|
|
25
|
Szegezdi E, Fitzgerald U and Samali A:
Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann
NY Acad Sci. 1010:186–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Urano F, Wang X, Bertolotti A, et al:
Coupling of stress in the ER to activation of JNK protein kinases
by transmembrane protein kinase IRE1. Science. 287:664–666. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Forrester JS and Libby P: The inflammation
hypothesis and its potential relevance to statin therapy. Am J
Cardiol. 99:732–738. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tournier C, Hess P, Yang DD, et al:
Requirement of JNK for stress-induced activation of the cytochrome
c-mediated death pathway. Science. 288:870–874. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuwana T, Mackey MR, Perkins G, et al:
Bid, Bax, and lipids cooperate to form supramolecular openings in
the outer mitochondrial membrane. Cell. 111:331–342. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reddy R, Chahoud G and Mehta JL:
Modulation of cardiovascular remodeling with statins: fact or
fiction? Curr Vasc Pharmacol. 3:69–79. 2005. View Article : Google Scholar : PubMed/NCBI
|

















