Introduction
Chromosomal abnormalities are hereditary diseases.
Approximately 95% of patients with chromosomal abnormalities suffer
the number variation of chromosomes 13, 18, 21, X and Y (1). The probability of trisomy 21 syndrome
is ~1/800 and the predominant symptoms for patients include
abnormal appearance and development, and a low IQ; the probability
of trisomy 18 is between 1/3,000 and 1/8,000, and the survival rate
is <1 year for the majority of patients; the probability of
trisomy 13 is ~1/2 that of trisomy 18 and the average life
expectancy of patients is 3 months (2–4). A
number of X and Y chromosome abnormalities result in developmental
and reproductive disorders. The case numbers of sex chromosome
abnormalities diagnosed in the Laboratory of Medical Genetics
(Maternal and Child Health Care Hospital of Qinzhou, Qinzhou,
China) are between those of trisomy 21 and those of trisomy 18.
Karyotype analysis, a common diagnostic test for chromosomal
abnormalities, typically takes 2–4 weeks. Due to the complex
operation of the cell culture and karyotype analysis, the risk of
failure of the amniotic fluid culture and time taken to obtain
results, karyotype analysis is limited in prenatal diagnosis.
Short tandem repeats (STRs) are genetic markers with
genetic polymorphism. They are composed of a number of variations
in the core sequence repeated units and widely exist in the human
genome. The majority of STRs with high polymorphism lie in
non-coding regions, free from selection pressure and have adequate
allelic products in gene amplification, thus, the markers have been
widely used in the fields of forensic medicine, human individual
identification and paternity testing (5,6). In
the quantitative fluorescence-polymerase chain reaction (QF-PCR)
test based on STR typing, the value of STR heterozygous loci of
normal individuals in the typing peak is closer to 1:1 while that
of STR loci in aneuploid chromosome trisomy in the peak is 2:1 or
1:1:1 (1–7). Thus, QF-PCR with the combination of
STR analysis may be used to detect chromosome copy numbers. QF-PCR
to analyze chromosome karyotype lasts ~24 h. However, misjudgment
risks may occur in diagnosing chromosome monomers if the use of STR
typing is the only program used to determine aneuploidy
diagnosis.
Homologous gene quantitative-PCR (HGQ-PCR) is a
method of detecting the ratio between the amplification sequence
and the gene copy number. It amplifies homologous nucleic acid
fragments with different lengths by using the same primers in
different chromosomes (8). For the
human chromosome genome, it is possible to diagnose the chromosomal
abnormality through its ratios in HGQ-PCR experiments, if the gene
fragment chosen only has one homologous sequence of the copy number
in a specific chromosome. If the homologous genes are between the
autosomes, the quantity of PCR product in the ratio of 1:1 is the
normal copy number, that in the ratio of 3:2 is the trisomy and
that in the ratio of 1:2 is the monomer. According to the above
methods, the HGQ-PCR experiment ratios of homologous sequences
between the autosomes and the sex chromosomes are analogized. Thus,
the fluorescence-labeled HGQ-PCR (fHGQ-PCR) has advantages in
chromosome monomer diagnosis.
TERF1 gene encodes the telomeric repeat-binding
factor 1 protein (9). Following
comparison by basic local alignment search tool (BLAST) of the
National Center for Biotechnology Information (NCBI), it was
observed that among homologous sequences of TERF1, a segment of
homologous sequences is located in chromosomes 13, 18 and 21. The
segment may be used to detect the copy numbers of chromosomes 13,
18 and 21 in fHGQ-PCR, thus it was termed 21-13-18-co1. In
addition, it was identified through the BLAST function provided by
the NCBI that a segment of homologous sequences are observed in
chromosome 18 and X. It is possible to detect the copy numbers of
chromosomes 18 and X by the amplification of the sequence, which we
termed X-18-co1. In the present study, 21-13-18-co1, X-18-co1 and
the gender loci Amelogenin (AMXY) which has been widely used in
forensic medicine were introduced. The situations of aneuploidy in
chromosomes 13, 18, 21 and X were determined by mutual
authentication with STR markers.
In the QF-PCR analysis, the direct labeling method
is usually used to label primers. In order to reduce the costs of
experimental labeling, 4 universal primers whose fluorescein is
labeled at the 5′ end respectively with 6-FAM, VIC, NED and PET
were designed. In this study, one end of the amplification primer
used the tailing method and tailed sequences were matched with
these 4 universal primers respectively. In the PCR amplification
test, the amplification primer and labeling primers were added into
the reaction tube together to achieve labeling of the amplification
product with fluorescence.
The purpose of this study was to develop a type of
QF-PCR reagent based on 4-color fluorescently labeled universal
primers combined with the fHGQ-PCR program. The reagent was able to
diagnose the aneuploidy in chromosomes 13, 18, 21, X and Y
accurately and rapidly, and met the requirements of screening
chromosome aneuploidies on a large scale.
Materials and methods
Materials
All experimental samples were obtained from DNA
samples stored in the Laboratory of Medical Genetics (Maternal and
Child Health Care Hospital of Qinzhou). Samples from 102 normal
people were collected by routine tests in the laboratory at random
for the pre-experimental study, to validate the method of fHGO-PCR.
Subsequent to the preliminary experiment, samples confirmed by
karyotype analysis were used for PCR amplification tests following
the extraction of DNA. In this study, 3 trisomy 21 samples, 2
trisomy 18 samples, 1 trisomy 13 sample, 3 aneuploidy in chromosome
X or Y samples and 10 normal samples were used to verify the QF-PCR
system. All aneuploidy samples were extracted from cultured
amniotic fluid cells, and all normal DNA samples were extracted
from uncultured blood cells. Lab-Aid DNA mini extraction kit
(Bio-V, Xiamen, Fujian, China) was used for DNA extraction. The
study was approved by the ethics committee of Maternal and Child
Health Care Hospital of Qinzhou and written informed consent was
obtained from the patients.
Design and verification of
multiple-tailed primers
An experiment was designed to verify the feasibility
of the QF-PCR based on the following universal primers:
AMXY-6-FAM-F (5′-6-FAM-CCC TGG GCT CT G TAA AGA ATA GTG-3′) and
CoA1-6-FAM (5′-6-FAM-CT C GAC ACG CAT CTG CTC AG-3′), whose
fluorescein are labeled at the 5′ end by directly using 6-FAM,
AMXY-R (5′-ATC AGA GCT TAA ACT GGG AAG CTG-3′) unlabeled and
CoA1-AMXY-F (5′-CTC GAC ACG CAT CTG CTC AG-CCC TGG GCT CTG TAA AGA
ATA GTG-3′) tailed as multiple-tailed primers. Four reaction tubes
were used in the method. The V1 reaction tube contained
AMXY-6-FAM-F and AMXY-R primers, and V2–V4 reaction tubes contained
CoA1-AMXY-F, AMXY-R and CoA1-6-FAM primers. The PCR reagent used
was 2X GoldStar Taq Mastermix (CoWin Biosciences, Inc., Beijing,
China) and the volume of the reaction liquid was 25 μl. The
reaction liquid contained 0.5 μl of every primer (10 μM) and 1 μl
DNA with the concentration of 50 ng/μl in male samples. PCR
amplification was conducted as follows: 95°C for 11 min, then a
variable number of cycles (V1 and V2, 28 cycles; V3, 30 cycles; V4,
32 cycles) of 95°C for 30 sec, 59°C for 30 sec and 72°C for 30 sec,
with a final extension at 72°C for 20 min. ABI 9700 (Applied
Biosystems, Foster City, CA, USA) was used to conduct PCR. Product
analysis was performed by ABI 3130 (Applied Biosystems), LIZ 500
(Applied Biosystems) was used as a size standard and degeneration
was conducted using deionized formamide (HiDi). In 3 analysis wells
in a 96-well plate, V1 products (0.3 μl) were added to each well
and combined with 0.3 μl V2–V4 products separately and the analysis
procedure began following mixing. A comparison of the quantity of
product of V1 and that of V2–V4 tubes was conducted. Subsequent to
this, the shape of the product of V1 was compared with that of the
product of V2–V4. Following verification, another 3 universal
primers termed CoA2 (5′-VIC-CTC GAC ACG CAT CTG ACG TT-3′), CoA3
(5′-NED-CTC GAC ACG CAT CTG GCG AA-3′) and CoA4 (5′-PET-CTC GAC ACG
CAT CTG CTA CC-3′) were designed. The primers were labeled at the
5′ end using VIC, NED and PET. CoA2, CoA3 and CoA4 were synthesized
by Life Technologies (Carlsbad, CA, USA), and the rest of the
primers were synthesized by Sangon (Shanghai, China).
Selection of STR loci
Based on the literature (10–14),
16 STR loci and the gender loci AMXY were selected to complete the
construction of the experimental system. STR loci with universal
primer CoA1, included D21S11, D18S1002, D21S2055 and D21S1412; STR
loci with universal primer CoA2 included D18S391, D18S51 and
D18S386; STR loci with universal primer CoA3, included AMXY D13S895
SRY, D13S631, D13S258, D21S1270 and D13S634; and STR loci with
universal primer CoA4, included DXS9902, X22 and XHPRT.
Design of fHGQ-PCR primers
According to the BLAST results, 21-13-18-co1 and
X-18-co1 multiple-tailed primers, the 21-13-18-co1 multiple-tailed
primer with universal primer CoA4 and the X-18-co1 multiple-tailed
primer with universal primer CoA3. The 21-13-18-co1 amplification
primers were as follows: Forward: 5′-CTC GAC ACG CAT CTG CTA CCA
GAC AGT AGG CTG CCT CAT G-3′ and reverse: 5′-GAA TAT CAT TTC TCA
GTC CCA AGC C-3′ for 21-13-18-co1; and forward: 5′-CTC GAC ACG CAT
CTG GCG AAT GGC CAA GGG GTT TAA CTC-3′ and reverse: 5′-CTT GTC TTT
CTT TAG GCC AAA T-3′ for 18-X-co1.
Single-tube QF-PCR system and
procedures
The single-tube QF-PCR system that was designed
included multiple-tailed primers of 16 STR loci, multiple-tailed
primers of AMXY, 21-13-18-co1, X-18-co1 and 4 universal primers
used for fluorescent labels. The PCR reagent used was 2X GoldStar
Taq Mastermix and the volume of the PCR reaction liquid was 25 μl.
The individual primer concentrations used in this system were as
follows: 0.4 μM D18S51, 0.12 μM D18S391, 0.32 μM D18S386, 0.2 μM
D21S2055, 0.4 μM D21S11, 0.16 μM D21S1412, 0.12 μM AMXY, 0.16 μM
D18S1002, 0.4 μM X-18-co1, 0.4 μM D13S895, 0.4 μM D13S631, 0.4 μM
D13S258, 0.4 μM D13S634, 0.4 μM D21S1270, 0.12 μM SRY, 0.2 μM
21-13-18-co1, 0.36 μM XHPRT, 0.4 μM X22 and 0.2 μM DXS9902. The
concentration of each fluorescently labeled universal primer was
0.4 μM and the template DNA was 50–100 ng. An ABI 9700
amplification instrument was used in the PCR experiment. PCR
amplification was conducted as follows: 95°C (11 min), then 29
cycles at 95°C (30 sec)/59°C (30 sec)/72°C (30 sec), and a final
extension at 72°C for 20 min. The products were analyzed using an
ABI 3130 genetic analyzer and the size standard was LIZ 500. Our
DNA samples were confirmed by karyotype analysis in the
laboratory.
Results
Results of multiple-tailed primers
In the QF-PCR experiments based on multiple-tailed
primers for demonstration, the results showed that the product peak
value of 30 reaction cycles with multiple-tailed reaction tubes is
similar with that of 28 reaction cycles with fluorescence-tailed
reaction tubes (Fig. 1). In the
present study, the hetero-peaks were not observed in the region
outside of the target band. Therefore, the QF-PCR experiment based
on universal primers is effective.
Validation of the method of fHGO-PCR
Among amplification products of 21-13-18-co1, the
amplification product of chromosome 21, 13 and 18 were 114, 120 and
121 bp, respectively. As there is only 1 bp difference between 120
and 121 bp, the two peaks in the peak figure tend to merge into one
in certain experiments. However, this does not affect the results,
as the ratio between 114 bp on the left and 120bp and 121bp on the
right approaches 1:1. When the DNA source is the entity of trisomy
21, the ratio between the left peak value and the right peak value
approaches 1.5:1. When the source of DNA is the entity of trisomy
13 or 18, the ratio between the left peak value and the right peak
value is closer to 1:1.5. The common validation with STR loci
distinguishes whether the samples exhibit trisomy 13 or 18.
Among the amplification products of X-18-co1, the
products for the X chromosome and chromosome 18 are 496 bp and 511
bp, respectively. In this locus amplification of DNA from normal
males, 496 bp represents the left products of the X chromosome,
while 511 bp is representative of the right products of chromosome
18. Thus, their ratios tend to be 1:2. However, in normal female
samples, the ratio between the left product peak value and the
right product peak value is close to 1:1. In male samples of
trisomy 18, the ratio between the left product peak value and the
right product peak value nearly approaches 1:3, while in the female
samples of trisomy 18 that increases to 2:3. This locus may also be
applied with AMXY to reduce the probablility of misjudgment from
STR markers diagnosing (45,X), (48,XXXX) and other sex chromosome
abnormalities. If AMXY only analyzes the X-peak and the ratio
between the X chromosome and chromosome 18 in the 18-X-co1 is 1:2,
the individual is (45,X); if AMXY simultaneously analyzes the
product peak at the ratio of 1:1 and the ratio between the X
chromosome and chromosome 18 in the 18-X-co1 is 1:1, the individual
is (48,XXYY). The diagnosis methods of other sex chromosome
anomalies are analogized based on the ratio association.
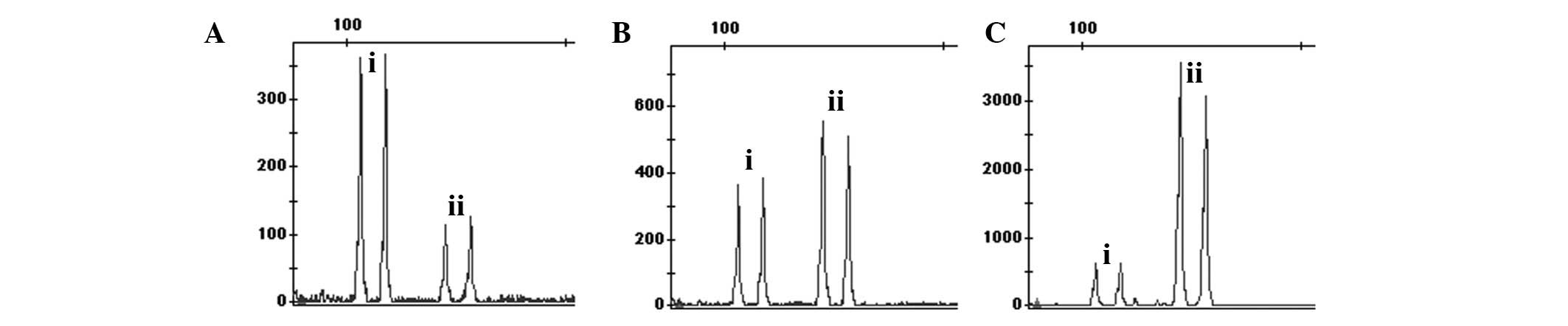
With the loci of AMXY (Fig. 2A), the ratio between the X peaks
and Y peaks is between 1.03 and 0.92 (male samples, X peak value as
1). With the loci of X-18-co1 (Fig.
2B), the ratio between the X peaks and chromosome 18 peaks is
between 2.27 and 1.50 (male samples, X peak value as 1) or between
1.31 and 1.04 (female samples, X peak value as 1). With the loci of
21-13-18-co1 (Fig. 2C), the ratio
between chromosome 21 peaks and chromosome 13/18 peaks is between
1.60 and 0.97 (chromosome 21 peak value as 1).
Results of the single-tube QF-PCR
system
The results (Fig.
3) of the single-tube QF-PCR system that was designed are
consistent with the results of the karyotype analysis in the
experiment of rapidly diagnosing the aneuploidy in chromosomes 13,
18, 21, X and Y.
Discussion
Studies have shown that the single-tube multiplex
QF-PCR is effective in the majority of diagnoses of the aneuploidy
in chromosomes 13, 18, 21, X and Y (1,2,10,12).
The majority of studies used the single-tube multiplex STR typing
technique which designs fluorescent labels to diagnose the
aneuploidy in chromosomes 13, 18, 21, X and Y through STR loci
typing results. However, there exist misjudgment risks in
diagnosing the aneuploidy in chromosomes 13, 18, 21, X and Y, and
the monomer in chromosomes 13, 18, 21, X and Y using the
single-tube multiplex STR typing technique alone. If labels from
chromosomes 13, 18, 21 or a chromosome relevant to chromosome X are
homozygous, testers will not be able to diagnose whether the
chromosome is a monomer by STR typing or these labels are only
produced by the same allele. Cirigliano et al(7) suggested Turner’s syndrome (45, X) and
other monomers may be diagnosed by the quantitative method with the
use of different fluorescence-labeled STR loci of similar size
fragments; however, there may also be a risk of misjudgment when
using this method as the fluorescence intensity varies with
different fluoresceins; it is debatable as it used different PCR
primers to quantify the amplification products of different loci,
as differences in the quantity of PCR product between similar
product length STR loci are not uncommon in day-to-day paternity
testing cases.
fHGQ-PCR is a method of detecting the copy number of
homologous genes. In the present study of fHGQ-PCR, AMXY gene loci
were used, which have been widely used in detecting genders of DNA
sequences obtained from individuals. Based on the principle of
fHGQ-PCR, this method may aid in increasing the reliability of
determining the quantity of the X chromosome by looking for the X
chromosome or the Y chromosome sequence homologous with autosomes.
In the present study 21-13-18-co1, X-18-co1 and AMXY loci were
introduced to diagnose aneuploidy in chromosomes 13, 18, 21, X and
Y. According to the results shown in Fig. 2, the ratio between the allele peaks
in AMXY or X-18-co1 vary compared with the perfect theoretical
ratio. Although the ratio between the allele peaks in 21-13-18-co1
are marginally higher than the theoretical ratio for the two peaks
for chromosome 13 and 18, and the peaks merge into one in certain
experiments, there remains a reference value in the diagnosis of
aneuploidy in chromosomes 13, 18 and 21 when the ratio between
chromosome 21 peaks and chromosome 13/18 peaks is >1.60 or
<0.8 (chromosome 21 peak value as 1), its effect is significant.
Nevertheless, Shadrach et al(15) and Roffey et al(16) respectively demonstrated the
misjudgment cases resulting from the gene loci AMXY mutation and a
study by Huang et al(17)
also confirmed the significant influence of genetic mutations in
PCR amplification efficiency. Moreover, the study of the
heterozygous amplification quantity of heterozygous alleles shows
the amplification quantity does not produce the theoretical ratio
but a ratio range (18).
Therefore, in the STR experiment, the quantity of the heterozygous
allele in normal individuals was not strictly 1:1 and the quantity
of the trisomy heterozygous gene was not strictly 1:1:1 or 1:2, but
varies compared with the perfect theoretical ratio. As the ratio
between genetic mutations and the quantity of product does not
follow the perfect theoretical ratio, there are misjudgment risks
in diagnosing chromosome aneuploidy using fHGQ-PCR alone.
This study concerning the comprehensive application
of STR amplification and fHGQ-PCR techniques achieves results
through mutual authentication between STR loci and fHGQ-PCR. It
neutralizes the disadvantages of simply using STR or fHGQ-PCR to
diagnose the aneuploidy in chromosomes 13, 18, 21, X and Y, and
ensures the reliability of the diagnosis results. Compared with the
2–4 weeks required by conventional karyotype analysis, using the
QF-PCR program in the present study markedly reduced the time, as
the program only takes 6 h to obtain the results when the DNA
samples have been extracted. In addition, the design of four-color
fluorescently labeled universal primers not only ensures the
accuracy of the experimental results, but also reduces the costs of
the QF-PCR system.
References
|
1
|
Ochshorn Y, Bar-Shira A, Jonish A and
Yaron Y: Rapid prenatal diagnosis of aneuploidy for chromosomes 21,
18, 13, and X by quantitative fluorescence polymerase chain
reaction. Fetal Diagn Ther. 21:326–331. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Andonova S, Vazharova R, Dimitrova V,
Mazneikova V, Toncheva D and Kremensky I: Introduction of the
QF-PCR analysis for the purposes of prenatal diagnosis in Bulgaria
- estimation of applicability of 6 STR markers on chromosomes 21
and 18. Prenat Diagn. 24:202–208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Palomaki GE, Deciu C, Kloza EM,
Lambert-Messerlian GM, Haddow JE, Neveux LM, et al: DNA sequencing
of maternal plasma reliably identifies trisomy 18 and trisomy 13 as
well as Down syndrome: an international collaborative study. Genet
Med. 14:296–305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin HY, Lin SP, Chen YJ, et al: Clinical
characteristics and survival of trisomy 18 in a medical center in
Taipei, 1988–2004. Am J Med Genet A. 140:945–951. 2006.PubMed/NCBI
|
|
5
|
Shin SH, Yu JS, Park SW, et al: Genetic
analysis of 18 X-linked short tandem repeat markers in Korean
population. Forensic Sci Int. 147:35–41. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim UK, Chae JJ, Lee SH, et al: Molecular
diagnosis of Duchenne/Becker muscular dystrophy by polymerase chain
reaction and microsatellite analysis. Mol Cells. 13:385–388.
2002.PubMed/NCBI
|
|
7
|
Cirigliano V, Ejarque M, Fuster C and
Adinolfi M: X chromosome dosage by quantitative fluorescent PCR and
rapid prenatal diagnosis of sex chromosome aneuploidies. Mol Hum
Reprod. 8:1042–1045. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee HH, Chang JG, Lin SP, Chao HT, Yang ML
and Ng HT: Rapid detection of trisomy 21 by homologous gene
quantitative PCR (HGQ-PCR). Hum Genet. 99:364–367. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen M, Haggblom C, Vogt M, Hunter T and
Lu KP: Characterization and cell cycle regulation of the related
human telomeric proteins Pin2 and TRF1 suggest a role in mitosis.
Proc Natl Acad Sci USA. 94:13618–13623. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Baig S, Ho SS, Ng BL, Chiu L, Koay ES,
Leow GH, et al: Development of quantitative-fluorescence polymerase
chain reaction for the rapid prenatal diagnosis of common
chromosomal aneuploidies in 1,000 samples in Singapore. Singapore
Med J. 51:343–348. 2010.
|
|
11
|
Liang W, Zhang L, Chen G, Xin J, Liao M
and Wu MY: Allele distributions for D21S1435 and D21S2055 loci in
two Chinese populations. J Forensic Sci. 47:667–668.
2002.PubMed/NCBI
|
|
12
|
Schmidt W, Jenderny J, Hecher K, Hackelöer
BJ, Kerber S, Kochhan L and Held KR: Detection of aneuploidy in
chromosomes X, Y, 13, 18 and 21 by QF-PCR in 662 selected
pregnancies at risk. Mol Hum Reprod. 6:855–860. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vats KR, Ishwad C, Singla I, Vats A,
Ferrell R, Ellis D, et al: A locus for renal malformations
including vesico-ureteric reflux on chromosome 13q33–34. J Am Soc
Nephrol. 17:1158–1167. 2006.PubMed/NCBI
|
|
14
|
Asmundo A, Perri F and Sapienza D: Allele
distribution of two X-chromosomal STR loci in a population from
Sicily (Southern Italy). International Congress Series.
1288:346–348. 2006. View Article : Google Scholar
|
|
15
|
Shadrach B, Commane M, Hren C and
Warshawsky I: A rare mutation in the primer binding region of the
amelogenin gene can interfere with gender identification. J Mol
Diagn. 6:401–405. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Roffey PE, Eckhoff CI and Kuhl JL: A rare
mutation in the amelogenin gene and its potential investigative
ramifications. J Forensic Sci. 45:1016–1019. 2000.PubMed/NCBI
|
|
17
|
Huang MM, Arnheim N and Goodman MF:
Extension of base mispairs by Taq DNA polymerase: implications for
single nucleotide discrimination in PCR. Nucleic Acids Res.
20:4567–4573. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leclair B, Frégeau CJ, Bowen KL and
Fourney RM: Systematic analysis of stutter percentages and allele
peak height and peak area ratios at heterozygous STR loci for
forensic casework and database samples. J Forensic Sci. 49:968–980.
2004. View Article : Google Scholar : PubMed/NCBI
|