Introduction
Heterogeneous Charcot-Marie-Tooth (CMT) neuropathy
is traditionally divided into a demyelinating or axonal type based
on the peripheral nerve conduction velocity of 38 m/sec on the
median nerve (1). A number of
genes are implicated in the demyelinating type (type I) whereas,
only a few are known for the axonal type (type II) (2,3). To
date, the most frequently reported mutated gene for axonal
neuropathy is the mitofusin-2 (MFN2) gene, and
it was first reported in 2004 (4).
The MFN2 mutations were found in ≤20% of HMSN II patients.
Phenotypes of axonal neuropathy due to a MFN2 mutation may
be divided in two groups; severe, early onset, which is more
frequent, usually associated with de novo mutations and
patients usually lose their ability to walk independently prior to
20 years of age. The late onset type is less frequent and usually
familial, associated with inherited dominant mutations.
The MFN2 gene encodes for mitochondrial
membrane protein, which plays a role in mitochondrial fusion and
fission (5). To date, ~110
mutations in the MFN2 gene are known (6). The current study reports the results
from diagnostic testing of 139 unrelated patients with axonal HMSN
neuropathy tested for MFN2 mutations. The clinical phenotype
of patients with mutations in the MFN2 gene is described and
the character of the novel mutations in the MFN2 gene is
discussed.
Patients and methods
A total of 139 Czech patients with sporadic or
familial presence of axonal neuropathy compatible with HMSN II were
selected for MFN2 testing. All examined patients signed
informed consent for DNA testing to clarify the cause of the
hereditary neuropathies and the study was approved by the Ethics
Committee of University Hospital Motol (Prague, Czech Republic).
The majority of the selected patients were previously tested
negative for the most common cause of CMT-CMT1A
duplication/hereditary neuropathy with liability to pressure
palsies deletion using the set of 17 microsatellite markers
(7), patients with late onset were
previously tested negative for mutations in the MPZ and
patients with early onset HMSN II were previously tested negative
for mutations in the GDAP1 gene.
All 17 coding exons and adjacent intron sequences
were amplified in 12 PCR fragments with a set of primers (available
upon request) and annealing temperature of 65ºC. The sequencing
reaction was performed for 15 fragments using BigDye Terminator
v3.1 sequencing kit and products were analyzed on an ABI 3130
Genetic Analyzer (all Applied Biosystems, Inc., Foster City, CA,
USA). The reference sequence used for MFN2 was NM_014874.3.
When the presence of a mutation was observed, samples from
additional family members were analyzed to evaluate the segregation
of the mutation with the disease.
When the mutation was observed, the prediction
programs SIFT (8) and PolyPhen-2
(9) were used for the
pathogenicity classification of the mutations. Exome Variant Server
(EVS) (10), 1000 Genomes
(11,12) and Single Nucleotide Polymorphisms
database (dbSNP) (13,14) were used to find information on the
variants. The EVS operates with more than 6,500 samples analyzed
for the exome variants, the current group was assumed to be large
enough and therefore the 100 DNA control samples were not analyzed
as was usual previously.
In addition, 73 patients from 64 families,
previously tested negative for the MFN2 mutations, were
examined with the multiplex ligation-dependent probe amplification
analysis method (MRC Holland, Amsterdam, The Netherlands) to
exclude larger rearrangements (duplications, deletions) not
detectable with Sanger sequencing. The SALSA MLPA kit,
P143-MFN2-MPZ was used and analysis was performed according to the
manufacturer’s instructions. Products were analyzed on an ABI 310
Genetic Analyzer. GeneMapper software v4.0 and Coffalyser v1.0.0.43
(MRC Holland) were used to analyze the data.
Results
A total of 11 nonsynonymous heterozygous mutations
were detected by sequencing in 15 unrelated patients from a group
of 139 patients with axonal neuropathy (Fig. 1). A total of eight were pathogenic,
the remaining three were observed in healthy relatives and are
therefore considered as rare benign polymorphisms (Table I). The frequency of detected
pathogenic MFN2 mutations in the cohort was 7.2%. The
prediction programs support the present observations, with the
exception of two cases; p.L209Q was classified as pathogenic, but
was also observed in a healthy relative and p.W740S was scored by
PolyPhen-2 as benign but was already reported as disease causing
(4,15). Five from the 11 reported mutations
are novel. No exonic deletion or duplication of any of the 17 exons
examined was detected with MLPA in the 73 patients from 64
families. A large number of previously reported polymorphisms was
observed. An overview of all observed polymorphisms with minor
allele frequency is summarized in Table II.
 | Table ISummary of the detected mutations and
results of predicting programs. |
Table I
Summary of the detected mutations and
results of predicting programs.
| Mutation | Amino acid
change | Nucleotide
change | No. of exon | Mutation
character | Novel/reported | dbSNP | EVS (f in %) | SIFT | PolyPhen-2 |
|---|
| 1 | p.E65X | c.193G>T | 4 | Pathogenic | Novel | - | - | No prediction | No prediction |
| 2 | p.R94W | c.280C>T | 4 | Pathogenic | Reported | rs119103263 | - | Deleterious | Potentially
damaging |
| 3 | p.T105R | c.314C>G | 5 | Pathogenic | Novel | - | - | Deleterious | Potentially
damaging |
| 4 | p.H165Y | c.493C>T | 6 | Pathogenic | Reported | - | - | Deleterious | Potentially
damaging |
| 5 | p.A166T | c.496G>A | 6 | Pathogenic | Reported | - | - | Deleterious | Potentially
damaging |
| 6 | p.L209E | c.626T>A | 7 | Benign | Novel | - | - | Deleterious | Potentially
damaging |
| 7 | p.C281S | c.842G>C | 9 | Benign | Novel | rs147136530 | 0.0077 | Tolerated | Benign |
| 8 | p.A485T | c.1453G>A | 14 | Unresolved | Novel | - | - | Deleterious | Benign |
| 9 | p.V705I | c.2113G>A | 18 | Benign | Reported | rs142271930 | 0.269 | Tolerated | Benign |
| 10 | p.R707W | c.2119C>T | 18 | Pathogenic | Reported | rs119103267 | 0.384 | Deleterious | Potentially
damaging |
| 11 | p.W740S | c.2219G>C | 19 | Pathogenic | Reported | rs28940292 | - | Deleterious | Benign |
| Table IISummary of detected polymorphisms
with known MAF and rs numbers. |
Table II
Summary of detected polymorphisms
with known MAF and rs numbers.
| Position | Amino acid
change | MAF | dbSNP |
|---|
| c.176-76G>A | - | NA | NA |
| c.474+65C>T | - | C=0.366 | rs2236056 |
| c.600-25T>C | - | C=0.058 | rs41278626 |
| c.709-32C>T | - | T=0.0009 | rs41278628 |
| c.870 C>T | p.Gly290Gly | NA | NA |
| c.957C>T | p.Gly319Gly | TT=0.004 | rs41278632 |
|
c.1039-22T>C | - | C=0.044 | rs6680984 |
|
c.1496-34T>C | - | C=0.008 | rs143065633 |
| c.1641C>T | p.Leu547Leu | NA | rs140924661 |
|
c.1872+63T>C | - | C=0.047 | rs2273295 |
| c.1873-66T-G | - | T=0.469 | rs7550536 |
|
c.2204+15T>C | - | C=0.083 | rs77262016 |
| c.*187C>G | - | NA | NA |
| c.*231G>A | - | A=0.011 | rs41278636 |
| c.*58A>G | - | A=0.305 | rs1042842 |
Families with pathogenic mutations
A novel pathogenic mutation, p.E65X, was observed in
a mother and her daughter. The mother was examined at 58 years of
age; the patient was capable of walking without support and only
mild foot deformity and arched instep were present. The daughter
exhibited the first signs of CMT in childhood and subsequently
underwent three orthopedic corrective surgeries on the both feet.
At the age of 29, the patient is capable of walking without
support, but has muscle atrophies below the knees and on the upper
limbs.
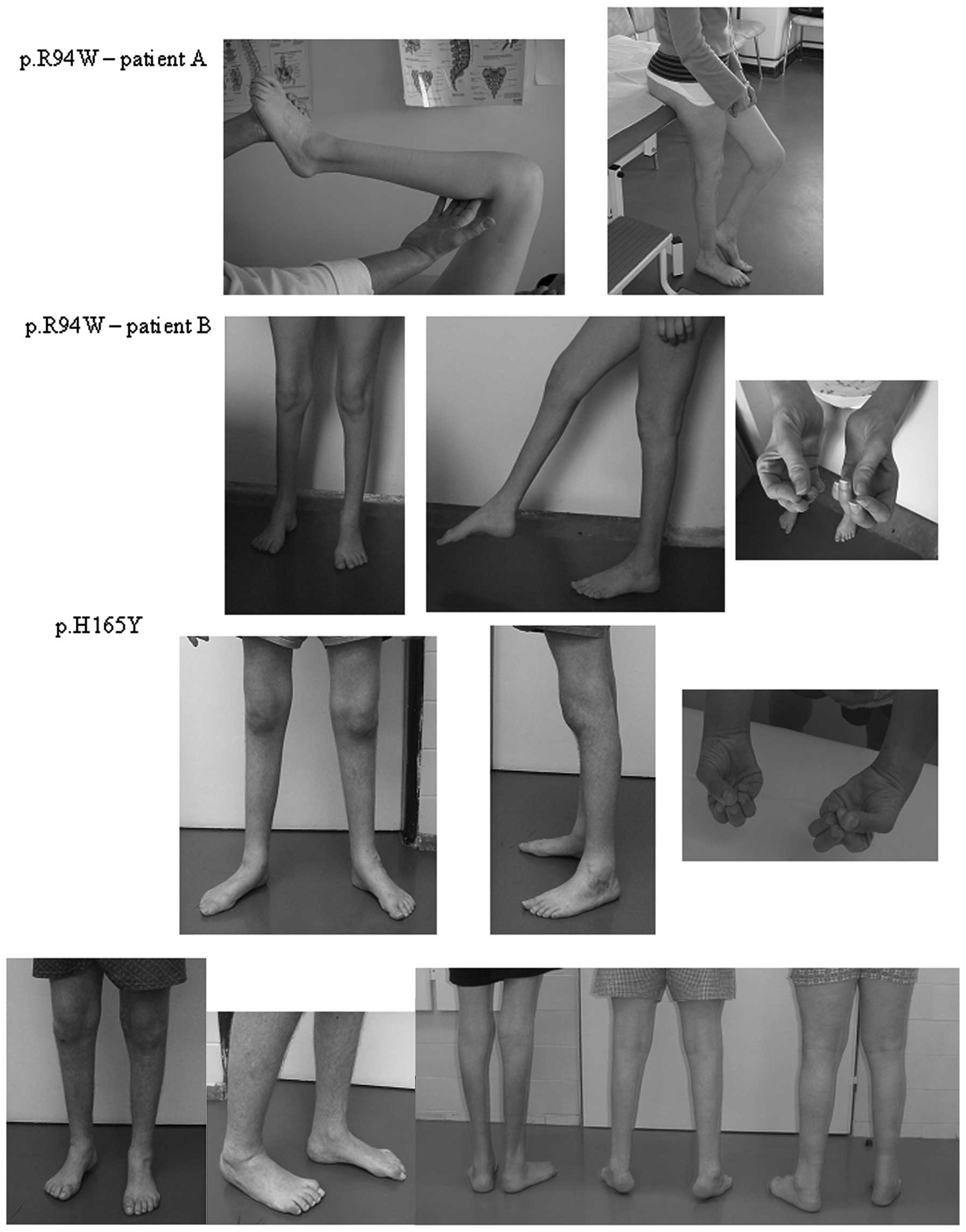
The mutation p.R94W, was observed in two patients
from different families. Disease onset was early, ~4-5 years of age
in the patients and the two were severely affected. The two
patients were ~30 years of age and suffer from pronounced muscle
atrophies extending above the knees and severe atrophies of the
small hand muscles were present (Fig.
2). Both patients had gait problems and required crutches.
Muscle strength was reduced in the wrist to 2/2 (MRC) in patient 2A
and 3/3 in patient 2B. Muscle strength in the ankles was reduced to
0 in both patients.
Mutation p.T105R was observed in two sisters
affected by axonal CMT neuropathy. The older sibling was more
severely affected, with the affliction beginning at 4 years of age.
From the age of 10 years, the patient had been using a wheelchair.
Following surgical prolongation of the Achilles tendons, the
patient is capable of walking with the support of canes. At the
current age of 28 years, the patient exhibits severe atrophies
above the knees and marked scoliosis. In her younger sister, the
disease manifested at the age of 14 years. At the current age of 23
years, the patient exhibits mild atrophies distally in the legs and
pes cavus deformities. The two sisters are heterozygotes for the
same mutation and it was not observed in their parents. Examination
using 17 microsatellite markers proved correct parenthood for the
parents, thus, a gonadal mosaicism in one of the parents is the
most probable explanation.
The known mutation p.H165Y was detected in two
families with similar phenotypes. In the two families, the disease
manifested early, ~6-10 years of age. The father (47 years) and
daughter (26 years) from the first family were severely affected.
They underwent a number of orthopedic corrective surgeries on their
feet and are capable of walking with the support of canes. The
patients have minor atrophies of the small hand muscles. The father
(54 years of age) and two sons (27 and 29 years of age) are
affected in the second family (Fig.
2). The father and older son had a number of orthopedic
corrective surgeries on their feet. The older brother is more
severely affected compared with the father and has atrophies of the
small hand muscles. The affected males from the second family are
capable of walking without support.
Mutation p.A166T was observed in a mother and son
who are mildly affected. The mother has had difficulties with
walking since childhood and the last examination at the age of 51
years revealed distal atrophies of the leg muscles, foot
deformities and shortened Achilles tendons. The son was examined at
the age of 34 years and clinically exhibited no manifestations of
CMT neuropathy. Axonal neuropathy was observed upon
electrophysiological examination.
Mutation p.R707W was observed in a 14-years old
patient with axonal neuropathy in whom symptoms of CMT manifested
~7 years of age. The patient exhibited muscle atrophies of the
lower limbs and atrophies of the small hand muscles. The patient’s
mother feels healthy and is declared as unaffected, but has the
same mutation. However, the mother denied any clinical or
electrophysiological examination.
Mutation p.W740S was observed in an affected patient
and his daughter. The patient’s sister, mother and uncle, had
similar difficulties but did not agree to genetic testing. The
patient has muscle atrophies of the lower limbs and foot
deformities. The daughter was clinically examined at 5 years of
age. The patient stumbles and has flat feet. Electrophysiological
examination was refused by the parents.
Family with a probable pathogenic
mutation
Mutation p.A485T was observed in a single patient
with axonal neuropathy which manifested at the age of 55 years.
Electrophysiological examination confirmed the axonal neuropathy.
The patient is currently 70 years old and has no children for
testing of segregation in the family.
Families with probable benign
variants
Mutations p.L209E and p.C281S were observed in three
families with axonal HMSN. The same mutations were also observed in
their healthy relatives, but clinical and electrophysiological
examination revealed no signs of CMT neuropathy. Mutation p.V705I
was observed in two patients from two unrelated families. The 70
years old patient in the first family has presented with problems
since childhood and his clinical phenotype is similar to CMT1A, but
exhibits axonal neuropathy according to electrophysiological
examination. A patient from the second family has had problems from
33 years of age. The patient has complicated diabetes mellitus and
after the neurological and electrophysiological examination the
HMSN is suspected.
Within this cohort with pathogenic mutations two
clusters of phenotypes were observed. i) Severe phenotype with age
of onset occurring at 4-6 years but with normal early motor
development and walking beginning at ~12 months of age was observed
more frequently (6 of 10 pathogenic mutations, 60%). Patients with
this phenotype usually lost their ability of independent walking
prior to the age of 20 years and require crutches or a wheelchair.
These patients are usually sporadic cases in the families and are a
result of a mutation which arose de novo. The mutations with
the de novo origin causing a severe phenotype are p.R94W and
p.T105R. The familial cases are p.H165Y and p.W740S. ii) The second
phenotype is mild with the age of onset between the 2nd and 5th
decade without loss of independent walking. This phenotype is
usually in families with more affected members. This phenotype was
observed in patients with p.E65X, p.A166T and p.R707W
mutations.
Discussion
Pathogenic mutations were detected in 7.2% (139/10)
of unrelated Czech Charcot-Marie-Tooth neuropathy type 2 (CMT2)
patients. Compared with Casasnovas et al(16) and Feely et al(17) who reported 16% for the Spanish
population and 21% for US and UK patients, the lower percentage in
the current study may be due to less stringent selection criteria
that was used and inclusion of more patients with late onset axonal
neuropathy and also due our to participation in a previous
collaborative study in 2006 (15),
where unresolved severely affected HMSN II patients were selected
and in 6 patients of 42, the pathogenic mutation was detected.
A number of mutations detected in the current
patient cohort in this study have previously been reported, namely;
p.R94W, p.H165Y, p.A166T, p.V705I, p.R707W and p.W740S. Two
patients with the p.R94W mutation are early and severely affected
with HMSN II but no other clinical signs, including optic atrophy
or increased tendon reflexes were observed. The same was reported
for this mutation in a number of other studies (15-20).
Only one family with p.R94W mutation developed optic atrophy at
ages 40 and 50 years (21).
The previously reported p.H165Y mutation was
observed in a Czech family (15).
It is highly probable that this family is related to the family
reported in the present study since they have the same family name,
however, it was not possible to prove this. Three other amino acid
changes were reported for the same codon p.H165 in HMSN II
patients; p.H165D in one family (22), p.H165R in two single patients
(15,19) and one family with additional signs
of tremor and sensorineural hearing loss (18) and p.H165L in a patient with mild to
moderate predominantly motor CMT neuropathy up to the age of 60
years, followed by progression acceleration, proximal limb muscle
weakness and bulbar involvement (23). The four substitutions showed that
this codon, p.H165, is a mutation hotspot in the MFN2 gene,
similar to p.R94 and others.
The p.A166T mutation was previously reported in a
family where the mother is wheelchair-bound and unable to write at
the age of 49 years. Her daughter exhibited distal muscle atrophies
in all four limbs at the age of 28 years (24). The clinical findings are similar to
the current observations where a 51-year-old mother is more
severely affected compared with her son at age 34 years where CMT
neuropathy was observed only upon electromyography examination.
The p.V705I mutation was previously reported as
causing disease in one patient with CMT2 (25). This mutation was observed in two
unrelated CMT2 patients. However, the SIFT and PolyPhen-2 scored it
as benign and the frequency for mutated allele in EVS is 0.3%. The
mutation is therefore potentially a polymorphism and is not causal
for CMT2 neuropathy in these patients or it may be a recessive
mutation.
The mild phenotype in patients with the p.W740S
mutation is similar to the phenotype previously reported for this
mutation (4,15,17).
In the MFN2 gene eight recessive mutations
have been reported. The first compound heterozygous mutation was
reported by Verhoeven et al(15), but without data from their parents.
Nicholson et al(26)
reported two compound heterozygotes and homozygous p.R707W in one
patient where all parents were asymptomatic carriers of
heterozygous mutations with minimal clinical findings of
neuropathy. Other compound heterozygous mutations, p.G108R and
p.R707W, were reported in three siblings by Calvo et
al(20) where the parents had
no signs of peripheral neuropathy, had normal electrophysiological
findings and each carried a single heterozygous mutation. Polke
et al(27) reported three
patients with recessive mutations. In two siblings the combination
of missense mutations and deletion of 2 exons was observed. A
heterozygous p.R707W mutation was detected in one patient with
peripheral neuropathy and also in her asymptomatic mother in this
study. The second mutation was not detected by sequencing, however
it is possible that it may be missed in a specific deep intronic
part, thus, the causal effect of recessive p.R707W mutation remains
possible.
Two of the five novel mutations are pathogenic. For
p.G65X no prediction was possible, however, it is a stop mutation.
p.G65X segregates in the family with the HMSN II phenotype and this
variant is not present in EVS. The second pathogenic mutation,
p.T105R, was detected in two sisters but not in their healthy
parents with confirmed parentity, thus a gonadal mosaicism in one
of the parents appears to be the most probable explanation. This
mutation is not observed in EVS but another amino acid change,
p.T105M, in the same codon was reported as causal and supports the
current conclusion (17,18,28).
Two other mutations, p.L209E and p.C281S, are assumed to be benign
variants or recessive mutations since they were also identified in
healthy relatives with normal findings under electrophysiological
examination.
The remaining novel mutation, p.A485T, was observed
in a single patient, thus, the pathogenicity may not be clearly
resolved. Moreover, the prediction by SIFT and PolyPhen-2 is
tolerated and benign and the variant was not detected in EVS. For a
more reliable prediction, another family with segregation of the
mutation together with the phenotype is required.
During examination of the Czech HMSN II patients for
MFN2 mutations 11 heterozygous mutations were observed, five
of them novel and six previously reported. The authors propose to
examine the MFN2 gene primarily in patients with early and
severe axonal CMT, where early motor development was normal, but
where patients have pronounced distal weakness in preschool age and
in patients with later onset of CMT2 between the 2nd and 5th decade
with autosomal dominant inheritance in the family. MLPA examination
did not reveal any duplication or deletion in MFN2 exons and
these aberrations are not a relevant cause of CMT. Performing MPLA
examination of the MFN2 gene in routine MFN2 gene
testing is therefore neither effective nor rational.
Acknowledgements
The study was supported by grant no. IGA MH CZ NS
10554-3 and by MH CZ – DRO, University Hospital Motol, Prague,
Czech Republic 00064203.
References
|
1
|
Harding AE and Thomas PK: The clinical
features of hereditary motor and sensory neuropathy types I and II.
Brain. 103:259–280. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Inherited Peripheral Neuropathies Mutation
Database. http://www.molgen.ua.ac.be/CMTMutations/Home/IPN.cfm.
Accessed October 3, 2013
|
|
3
|
Züchner S and Vance JM: Molecular genetics
of autosomal-dominant axonal Charcot-Marie-Tooth disease.
Neuromolecular Med. 8:63–74. 2006.PubMed/NCBI
|
|
4
|
Züchner S, Mersiyanova IV, Muglia M, et
al: Mutations in the mitochondrial GTPase mitofusin 2 cause
Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 36:449–451.
2004.PubMed/NCBI
|
|
5
|
Nunnari J, Marshall WF, Straight A, Murray
A, Sedat JW and Walter P: Mitochondrial transmission during mating
in Saccharomyces cerevisiae is determined by mitochondrial
fusion and fission and the intramitochondrial segregation of
mitochondrial DNA. Mol Biol Cell. 8:1233–1242. 1997.PubMed/NCBI
|
|
6
|
Stenson PD, Mort M, Ball EV, et al: The
Human Gene Mutation Database: 2008 update. Genome Med. 1:132009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Seeman P, Mazanec R, Zidar J, Hrusáková S,
Ctvrtecková M and Rautenstrauss B: Charcot-Marie-Tooth disease type
1A (CMT1A) and hereditary neuropathy with liability to pressure
palsies (HNPP): reliable detection of the CMT1A duplication and
HNPP deletion using 8 microsatellite markers in 2 multiplex PCRs.
Int J Mol Med. 6:421–426. 2000.
|
|
8
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome Res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sunyaev S, Ramensky V, Koch I, Lathe W
3rd, Kondrashov AS and Bork P: Prediction of deleterious human
alleles. Hum Mol Genet. 10:591–597. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
NHLBI Exome Sequencing Project (ESP).
Exome Variant Server (EVS). http://evs.gs.washington.edu/EVS.
Accessed October 3, 2013
|
|
11
|
Abecasis GR, Altshuler D, Auton A, et al;
1000 Genomes Project Consortium. A map of human genome variation
from population-scale sequencing. Nature. 467:1061–1073. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
1000 Genomes. http://www.1000genomes.org.
Accessed October 3, 2013
|
|
13
|
Sherry ST, Ward MH, Kholodov M, et al:
dbSNP: the NCBI database of genetic variation. Nucleic Acids Res.
29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
National Center for Biotechnology
Information, National Library of Medicine. Database of Single
Nucleotide Polymorphisms (dbSNP). http://www.ncbi.nlm.nih.gov/SNP.
Accessed October 3, 2013
|
|
15
|
Verhoeven K, Claeys KG, Zuchner S, et al:
MFN2 mutation distribution and genotype/phenotype correlation in
Charcot-Marie-Tooth type 2. Brain. 129:2093–2102. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Casasnovas C, Banchs I, Cassereau J, et
al: Phenotypic spectrum of MFN2 mutations in the Spanish
population. J Med Genet. 47:249–256. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Feely SM, Laura M, Siskind CE, et al: MFN2
mutations cause severe phenotypes in most patients with CMT2A.
Neurology. 76:1690–1696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chung KW, Kim SB, Park KD, et al: Early
onset severe and late-onset mild Charcot-Marie-Tooth disease with
mitofusin 2 (MFN2) mutations. Brain. 129:2103–2118. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cho HJ, Sung DH, Kim BJ and Ki CS:
Mitochondrial GTPase mitofusin 2 mutations in Korean patients with
Charcot-Marie-Tooth neuropathy type 2. Clin Genet. 71:267–272.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Calvo J, Funalot B, Ouvrier RA, et al:
Genotype-phenotype correlations in Charcot-Marie-Tooth disease type
2 caused by mitofusin 2 mutations. Arch Neurol. 66:1511–1516. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Züchner S, De Jonghe P, Jordanova A, et
al: Axonal neuropathy with optic atrophy is caused by mutations in
mitofusin 2. Ann Neurol. 59:276–281. 2006.PubMed/NCBI
|
|
22
|
Zhu D, Kennerson ML, Walizada G, Züchner
S, Vance JM and Nicholson GA: Charcot-Marie-Tooth with pyramidal
signs is genetically heterogeneous: families with and without MFN2
mutations. Neurology. 65:496–497. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marchesi C, Ciano C, Salsano E, et al:
Co-occurrence of amyotrophic lateral sclerosis and
Charcot-Marie-Tooth disease type 2A in a patient with a novel
mutation in the mitofusin-2 gene. Neuromuscul Disord. 21:129–131.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Loiseau D, Chevrollier A, Verny C, et al:
Mitochondrial coupling defect in Charcot-Marie-Tooth type 2A
disease. Ann Neurol. 61:315–323. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Engelfried K, Vorgerd M, Hagedorn M, et
al: Charcot-Marie-Tooth neuropathy type 2A: novel mutations in the
mitofusin 2 gene (MFN2). BMC Med Genet. 7:532006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nicholson GA, Magdelaine C, Zhu D, et al:
Severe early-onset axonal neuropathy with homozygous and compound
heterozygous MFN2 mutations. Neurology. 70:1678–1681. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Polke JM, Laurá M, Pareyson D, et al:
Recessive axonal Charcot-Marie-Tooth disease due to compound
heterozygous mitofusin 2 mutations. Neurology. 77:168–173. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lawson VH, Graham BV and Flanigan KM:
Clinical and electrophysiologic features of CMT2A with mutations in
the mitofusin 2 gene. Neurology. 65:197–204. 2005. View Article : Google Scholar : PubMed/NCBI
|