Introduction
Lysophosphatidic acid (LPA) affects various cell
functions, including cell migration, proliferation, differentiation
and survival, through a family of cognate G-protein coupled
receptors (GPCRs). The binding of LPA to its cognate receptors
elicits a range of LPA downstream signaling through at least three
different types of G proteins, including Gi/o, Gq/11 and G12/13
(1). LPA is also involved in
diverse neurodevelopmental processes, such as neurogenesis,
neuritogenesis and neuronal migration. We have previously
demonstrated that LPA protected differentiating hippocampal
progenitor cells (HPCs) from apoptosis by inhibiting the
phosphorylation of glycogen synthase kinase 3 (GSK3) (2), suggesting that LPA may be involved in
hippocampal neurogenesis. GSK3 plays a central role in the
canonical Wnt/β-catenin signaling pathway, which is shown to be
important in neurogenesis by supporting the proliferation of neural
stem and precursor cells (3).
Previous studies have demonstrated novel aspects of Wnt/β-catenin
signaling in neurogenesis. Wnt/β-catenin signaling is involved in
neurogenesis by regulating the differentiation of neural precursors
(4–7). Moreover, studies have revealed that
β-catenin signaling promotes the survival of neuronal cells,
including precursor cells (8–12).
This has led to the hypothesis that β-catenin signaling may
regulate neurogenesis by modulating the survival of neuronal
progenitor cells (NPCs). LPA is also involved in the regulation of
cortical neurogenesis by controlling the survival of NPCs in ex
vivo cortical culture systems, although the molecular mechanism
for this is not fully understood (13). Increasing data demonstrate that the
activation of G proteins alone, or the activation of GPCRs by
agonists such as lipid metabolites and growth factors, may induce
the stabilization of β-catenin and the subsequent β-catenin/T cell
factor (TCF) transcriptional activation that is independent of Wnt
(14,15).
In the present study, it was investigated whether
LPA induced β-catenin/TCF transcriptional activation and whether
β-catenin/TCF signaling by LPA was involved in the control of NPC
survival. It was demonstrated that LPA activated the β-catenin/TCF
signaling predominantly in Gi/o- and protein kinase C
(PKC)-dependent pathways, and that the β-catenin/TCF signaling
induced by LPA was involved in the suppression of apoptosis in
differentiating H19-7 cells. Therefore, the activation of the
β-catenin/TCF signaling pathway by LPA may function in neurogenesis
by controlling the survival of neural precursors.
Materials and methods
Materials
H19-7 cells were generously provided by Dr. Kwang C.
Chung (Yonsei University, Seoul, Republic of Korea). LPA
(1-oleoyl-2-hydroxy-sn-glycero-3-phosphate),
5-bromodeoxyuridine (BrdU), fatty acid-free bovine serum albumin
and pertussis toxin (PTX) were purchased from Sigma (St. Louis, MO,
USA). Wortmannin and GF109203 were purchased from Tocris Bioscience
(Bristol, UK). TOP Flash and FOP Flash luciferase reporters were
purchased from Upstate Biotechnology (Lake Placid, NY, USA). Alexa
Fluor 568-conjugated Annexin V was purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). Dulbecco’s modified Eagle’s
medium (DMEM), fetal bovine serum (FBS) and G418 were obtained from
Invitrogen Life Technologies. All other reagents were of analytical
grade or the highest purity available.
Cell culture
H19-7 cells, conditionally immortalized, embryonic
(day 17) rat hippocampal cells with a temperature-sensitive SV40
large T antigen (16), were
maintained at 33ºC (the permissive temperature at which SV40 large
T is functional) in DMEM supplemented with 10% FBS, 0.2 mg/ml G418,
50 U/ml penicillin and 50 mg/ml streptomycin. For differentiation,
H19-7 cells were shifted to 39ºC to eliminate the expression of the
functional large T antigen, then incubated for 24 h in DMEM
supplemented with N2 (N2 medium), and treated with LPA. The
differentiation of H19-7 cells into neuronal cells was confirmed by
the expression of neuronal markers, such as neurofilament-M and
neuron specific enolase, and cell morphology as previously
described (17). To determine
effect of pharmacological inhibitors on the TCF transcriptional
activation and inhibitory phosphorylation of GSK3β induced by LPA,
the H19-7 cells were pretreated with the inhibitor for 1 h before
LPA treatment.
Measurement of apoptosis by Annexin V
staining
Apoptosis was measured by staining with Annexin V
conjugated to Alexa Fluor 568 (Life Technologies, Grand Island, NY,
USA) as previously described (2).
H19-7 cells were collected, washed with phosphate-buffered saline
(PBS) and resuspended in 100 μl binding buffer [10 mM HEPES (pH
7.4), 140 mM NaCl and 2.5 mM CaCl2]. The cells were then
incubated with 5 μl Annexin V-Alexa Fluor 568 for 15 min at room
temperature in the dark, followed by the addition of 400 μl binding
buffer. The cells were analyzed by flow cytometry using a Guava
EasyCyte (GE Healthcare, Piscataway, NJ, USA).
Western blot analysis
H19-7 cells were collected by brief centrifugation
at 500 × g and resuspended in lysis buffer [10 mM HEPES (pH 7.5),
10 mM KCl, 1 mM EGTA, 1 mM EDTA, 1% Nonidet P-40, 1 mM
Na3VO4, 5 mM NaF and protease inhibitor
cocktail]. Following incubation on ice for 10 min, cell lysates
were centrifuged at 4,000 × g for 1 min. Supernatants containing
cytoplasm were collected and the remaining nuclear pellet was
resuspended in lysis buffer containing 0.1% sodium dodecyl sulphate
(SDS). The protein concentrations within each lysate were
determined by the Bradford assay (Bio-Rad, Richmond, CA, USA). The
fractionated extracts were mixed with SDS sample buffer and equal
quantities of the samples were loaded and separated by
SDS-polyacrylamide gel electrophoresis (8 or 10% reducing gels),
transferred to polyvinylidene difluoride membranes (Millipore,
Bedford, MA, USA) and blocked with 5% non-fat milk. The membranes
were incubated in primary antibody overnight at 4ºC. Subsequent to
this, the membranes were washed in TBST [10 mM Tris, 140 mM NaCl
and 0.1% Tween-20 (pH 7.6)], incubated with the appropriate
secondary antibody and washed again in TBST. Bands were visualized
by enhanced chemiluminescence and exposed to X-ray film.
Immunocytochemistry
Differentiating H19-7 cells cultivated on chamber
slides (Nalgene, Rochester, NY, USA) were treated with LPA. The
cells were fixed and permeabilized as described in a previous study
(2), processed for
immunocytochemistry with mouse anti-β-tublin III (Sigma) or rabbit
anti-β-catenin (Abcam, Cambridge, MA, USA), and subsequently
processed with Alexa Fluor 568- or Alexa Fluor 488-conjugated
secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA).
The stained cells were observed using a fluorescence microscope
(Axio Scope A1; Zeiss, Oberkochen, Germany) with a digital camera
attachment.
Luciferase reporter gene assay
H19-7 cells were transiently transfected with a
luciferase reporter and Renilla luciferase (pRL-TK; Promega
Corporation, Madisson, WI, USA) with the indicated plasmids, using
FuGene HD (Roche Molecular Biochemicals, Mannheim Germany). The
luciferase reporter constructs TOP Flash and FOP Flash (Upstate
Biotechnology) were used to determine the TCF/lymphoid enhancing
factor (LEF) transcription activity. Following transfection for 6
h, the cells were cultivated in N2 medium for 18 h and to induce
differentiation as described previously. The cells were treated
with LPA or vehicle for 4 h and collected. The cell extracts were
prepared by rinsing each plate twice with PBS and lysing the cells
in 150 μl Reporter Assay Lysis buffer (Promega Corporation). The
cell lysates were collected, normalized for protein content and
assayed using a dual luciferase assay system (Promega Corporation).
They were analyzed for firefly luciferase (TOP Flash or FOP Flash)
activity, and the reactions were quenched and immediately
reanalyzed for Renilla luciferase activity using an Autolumat
Luminometer (Berthhold Technologies, Oak Ridge, TN, USA).
Statistical analysis
Results are presented as the mean ± SEM. Statistical
significance was analyzed by one-way analysis of variance followed
by a Dunnett’s multiple test or a paired t-test using Prism
(GradPad Software, La Jolla, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
LPA induces activation of β-catenin/TCF
signaling in H19-7 cells
In a previous study, it was demonstrated that LPA
stimulated the inactivation of GSK-3 in H19-7 cells through
LPA1 and LPA2(2). As β-catenin is a substrate
for GSK3 and is degraded following phosphorylation by GSK3, and as
LPA activates G proteins, such as Gq, Gi/o and G12/13 (via coupling
to LPA1 and LPA2), LPA may activate
β-catenin/TCF signaling in H19-7 cells. To determine whether LPA
activated β-catenin/TCF signaling, cytoplasmic and nuclear levels
of β-catenin in the H19-7 cells treated with LPA were investigated
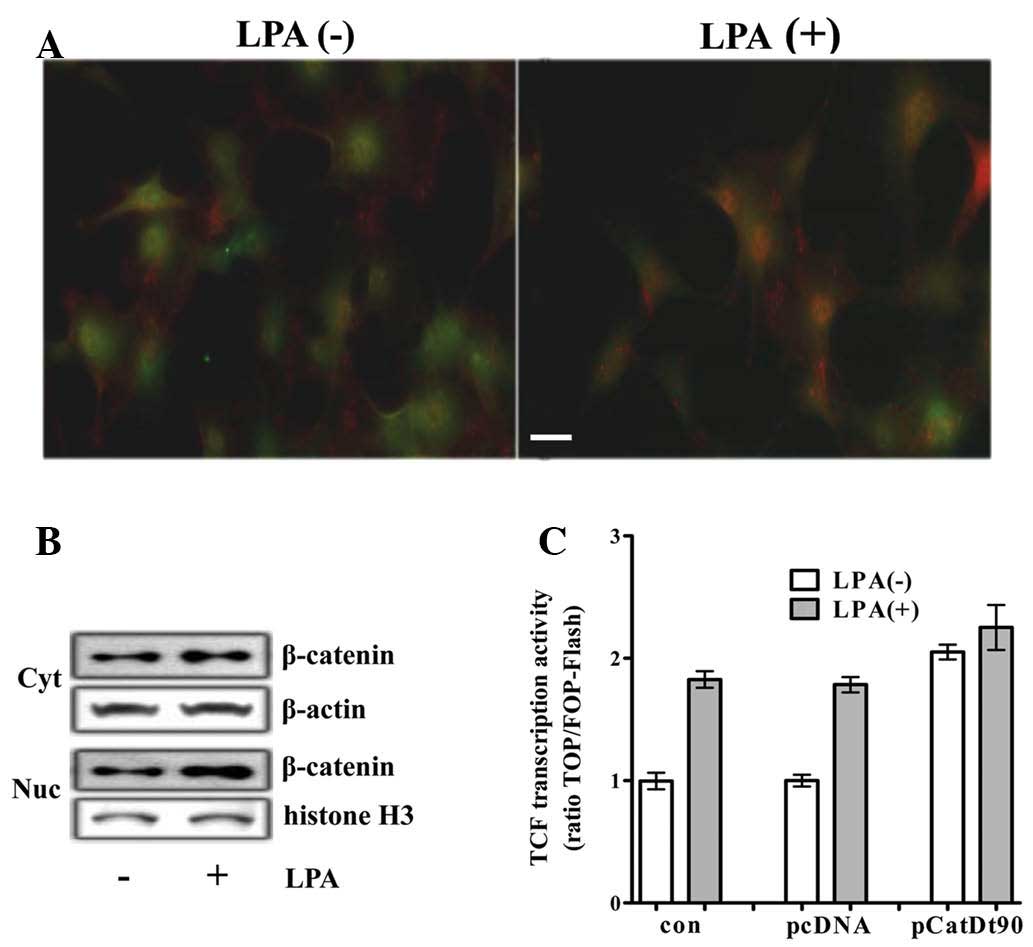
by immunocytochemistry and western blot analysis. An increase in
the level and the nuclear translocation of β-catenin was detected
by immunocytochemistry in the H19-7 cells (Fig. 1A). In addition, an increase in the
cytoplasmic levels of β-catenin and the subsequent increase in the
levels of nuclear β-catenin was demonstrated by western blot
analysis (Fig. 1B). An increase in
the level of nuclear β-catenin was also detected within 30 min
(data not shown). To determine whether LPA activated
β-catenin-mediated transcriptional activation, the TCF reporter
activity was analyzed in differentiating H19-7 cells transfected
with TCF luciferase reporter. TCF with LPA reporter activity was
~2-fold that without LPA, and was similar to that induced by
transfection with a plasmid encoding β-catenin truncated N-terminal
90 amino acids (b-CatΔ90) acting as a constitutively active mutant
of β-catenin (CA-β-catenin) (Fig.
1C).
LPA activates β-catenin/TCF signaling in
a PTX- and PKC-dependent manner
Our previous study demonstrated that LPA induced
cell survival in a PTX-dependent manner by the post-translational
upregulation of Mcl-1 through the inhibitory phosphorylation of
GSK3 in differentiating H19-7 cells. The inhibitory phosphorylation
of GSK3 is mediated predominantly by PKC and partly by mitogen
activated protein kinases, PKA and phosphatidylinositol 3-kinase
(PI3K) in the H19-7 cells (2). To
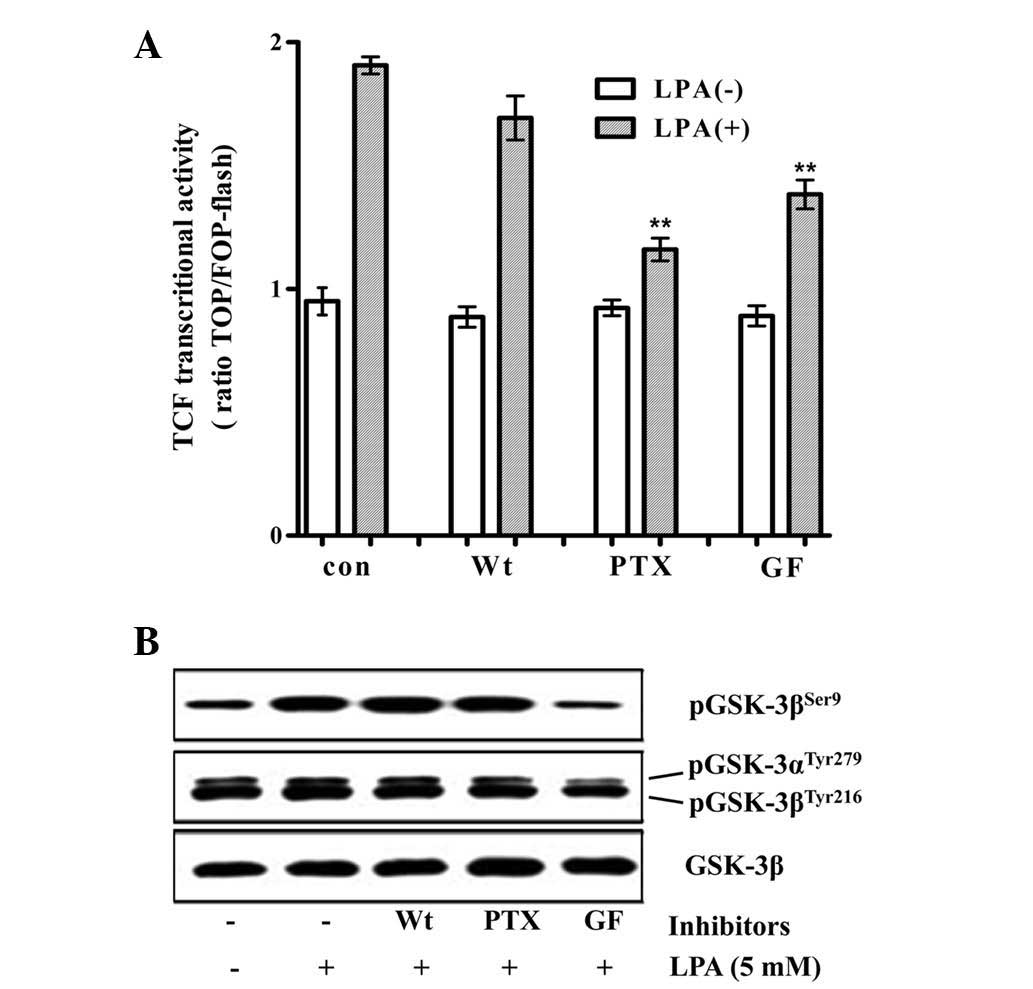
determine the mechanism by which LPA stimulates β-catenin/TCF
signaling, the effect of pharmacological inhibitors of PKC, PI3K
and Gi/o on the LPA-induced activation of the TCF reporter activity
and on the inhibitory phosphorylation of GSK3β at Ser9 by LPA was
analyzed. GF109203 (a PKC inhibitor) and PTX (a Gi/o specific
inhibitor) significantly decreased the LPA-induced activation of
TCF reporter activity, whereas wortmannin (a PI3K inhibitor) did
not significantly decrease this activation. By contrast, the
LPA-induced inhibitory phosphorylation of GSK3β was marginally
decreased by PTX and not by wortmannin, while GF109203 completely
inhibited the phosphorylation (Fig.
2). The observation that PTX significantly inhibited the
LPA-induced β-catenin/TCF signaling, with a marginal decrease in
the inhibitory phosphorylation of GSK3β, suggested that LPA may
stimulate β-catenin/TCF transactivation via signaling pathways that
are independent of the direct inhibition of GSK3.
Activation of β-catenin/TCF transcription
by LPA suppresses the apoptosis of H19-7 cells
LPA induced cell survival in a dose-responsive
manner in the differentiating H19-7 cells; however, the H19-7 cells
treated with LPA were not stained with BrdU, suggesting that LPA
induced cell survival by protecting H19-7 cells from cell death,
not by inducing cell proliferation (data not shown). To determine
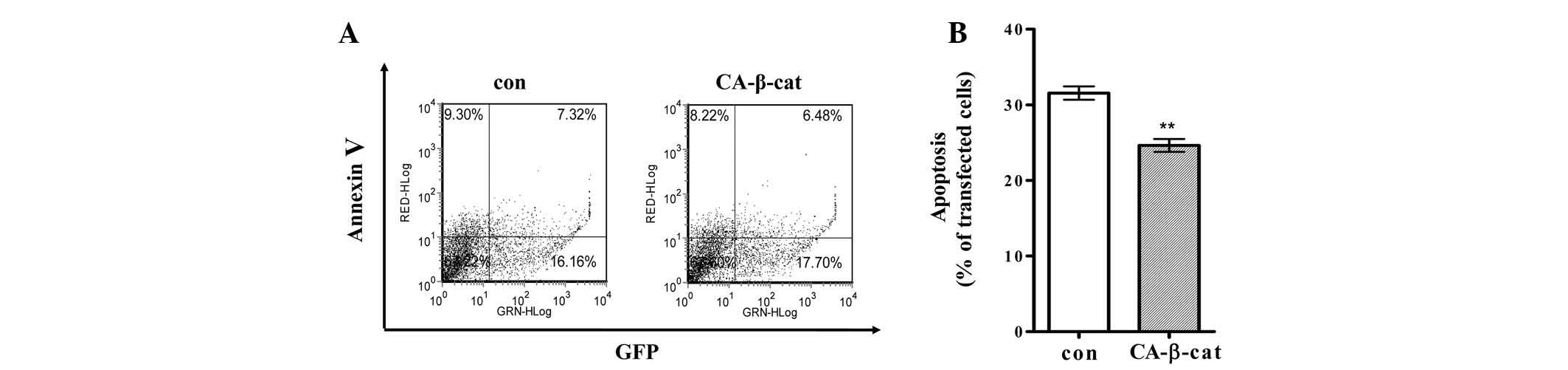
whether LPA-mediated β-catenin/TCF signaling was involved in
protecting H19-7 cells from cell death, the cells were transfected
with a plasmid expressing either CA-β-catenin or empty vector and a
plasmid expressing GFP. Transfected cells differentiated and
apoptosis was detected by flow cytometry as described in the
Materials and methods. Apoptosis was significantly suppressed in
the H19-7 cells expressing CA-β-catenin compared with that of the
control cells (Fig. 3).
Discussion
Activation of the cognate GPCRs by lipid
metabolites, growth factors or a P2Y receptor agonist was shown to
activate β-catenin/TCF signaling, although the mechanism of action
is unclear. The GPCR-mediated β-catenin/TCF transcriptional
activation (except for the P2Y receptor-mediated TCF activation)
was demonstrated to be implicated in oncogenicity, including cell
proliferation and epithelial-mesenchymal transition in cancer
cells, but not in normal physiological processes (18–23).
In the present study, it was demonstrated that LPA activated
β-catenin/TCF signaling predominantly via the Gi/o- and
PKC-dependent pathways in differentiating HPCs, and that the
β-catenin/TCF signaling induced by LPA was involved in suppressing
apoptosis in the HPCs. Activation of β-catenin/TCF signaling was
determined by an increase in the nuclear levels of β-catenin and
the stimulation of TCF reporter activity (Fig. 1). The observation that
β-catenin/TCF transactivation induced the expression of Mcl-1, an
anti-apoptotic Bcl2 family member (24), was consistent with a previous study
demonstrating that LPA-induced cell survival was mediated by
post-translational and transcriptional upregulation of Mcl-1
(2). Whether the direct inhibition
of GSK3β is involved in β-catenin stabilization and subsequent
β-catenin/TCF transactivation remains controversial. LPA was
previously shown to induce the inhibitory phosphorylation of GSK3β
by PKC, mitogen-activated protein kinase and protein kinase A, but
not PI3K, in H19-7 cells (2).
Therefore, in the present study, it was investigated whether
inhibitory phosphorylation of GSK3β by upstream kinases was
involved in the β-catenin/TCF transactivation. LPA-induced
activation of β-catenin/TCF signaling was significantly inhibited
by PTX, although PTX marginally decreased the inhibitory
phosphorylation of GSK3β. Moreover, the PKC inhibitor decreased the
β-catenin/TCF signaling less potently than that by PTX, although
the PKC inhibitor completely abolished the inhibitory
phosphorylation of GSK3β. Thus, these results suggested that LPA
may activate β-catenin/TCF transactivation via signaling pathways
that are independent of the direct inhibition of GSK3. However,
β-catenin/TCF transactivation may have occurred through direct
inhibition of GSK3β, as the PKC inhibitor significantly decreased
the activation of β-catenin/TCF signaling and the inhibitory
phosphorylation of GSK3β. Previous studies have suggested that
GSK3β only modifies β-catenin when GSK3 forms the
Axin/APC/β-catenin destructive complex (25,26),
and that only a small pool of GSK3β exists in the Axin scaffolding
complex within which GSK3β is not readily accessible to upstream
kinases (27). Thus, GPCR-mediated
activation of β-catenin/TCF signaling may occur without direct
inhibitory phosphorylation of GSK3β in the Axin scaffolding
complex, although upstream kinases are involved in this signaling
pathway and inhibitory phosphorylation of GSK3β in the other pool
is increased. Alternatively to the phosphorylation of GSK3 within
the Axin complex, the upstream kinases may activate β-catenin/TCF
signaling by acting on other targets involved in the activation of
β-catenin/TCF signaling, such as 14-3-3-mediated nuclear efflux of
β-catenin by Akt or PKC and KSRP-mediated stabilization of
β-catenin mRNA by PI3K/Akt (28–30).
Thus, PKC may activate β-catenin/TCF signaling without inhibitory
phosphorylation of GSK3β in the Axin complex, although the total
inhibitory phosphorylation of GSK3β is increased.
Expression of CA-β-catenin suppressed apoptosis in
differentiating H19-7 cells, suggesting that the activation of the
TCF reporter activity by LPA was involved in cell survival of HPCs.
Apoptosis of the H19-7 cells was suppressed by 6% following
expression of CA-β-catenin (Fig.
3). Suppressive effects of β-catenin/TCF activation on the
survival of the H19-7 cells were marginal, but may be
physiologically relevant, as small changes in the survival of
progenitor pools may produce a marked effect on neurodevelopment.
It was suggested that only a 3.5% increase in survival and a 2.5%
increase in terminal mitosis of progenitor cells induced an ~30%
increase in the thickness of the cerebral cortical wall (13). In concordance with the present
results, previous studies demonstrated that β-catenin/TCF signaling
was involved in the survival of neural precursors, although the
molecular mechanisms remain unknown (31–33).
In conclusion, the results suggested that LPA may regulate
β-catenin/TCF signaling in NPCs, as LPA is involved in neurogenesis
by controlling the survival of neural precursors.
Acknowledgements
This study was supported by the National Research
Foundation of Korea Grant funded by the Korean Government (grant
no. 2010-0005848), and by the Priority Research Centers Program
through the National Research Foundation of Korea (NRF), funded by
the Ministry of Education, Science and Technology (grant no.
NRF-2009-0094071).
References
|
1
|
Anliker B and Chun J: Lysophospholipid G
protein-coupled receptors. J Biol Chem. 279:20555–20558. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sun Y, Nam JS, Han DH, et al:
Lysophosphatidic acid induces upregulation of Mcl-1 and protects
apoptosis in a PTX-dependent manner in H19-7 cells. Cell Signal.
22:484–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang L, Yang X, Yang S and Zhang J: The
Wnt/β-catenin signaling pathway in the adult neurogenesis. Eur J
Neurosci. 33:1–8. 2011.
|
|
4
|
Ille F, Atanasoski S, Falk S, et al:
Wnt/BMP signal integration regulates the balance between
proliferation and differentiation of neuroepithelial cells in the
dorsal spinal cord. Dev Biol. 304:394–408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang J, Woodhead GJ, Swaminathan SK, et
al: Cortical neural precursors inhibit their own differentiation
via N-cadherin maintenance of beta-catenin signaling. Dev Cell.
18:472–479. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang X, Kopinke D, Lin J, et al: Wnt
signaling regulates postembryonic hypothalamic progenitor
differentiation. Dev Cell. 23:624–636. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang M, Villaescusa JC, Luo SX, et al:
Interactions of Wnt/beta-catenin signaling and sonic hedgehog
regulate the neurogenesis of ventral midbrain dopamine neurons. J
Neurosci. 30:9280–9291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pöschl J, Grammel D, Dorostkar MM,
Kretzschmar HA and Schüller U: Constitutive activation of β-catenin
in neural progenitors results in disrupted proliferation and
migration of neurons within the central nervous system. Dev Biol.
374:319–332. 2013.
|
|
9
|
Zhao S, Fu J, Liu X, Wang T, Zhang J and
Zhao Y: Activation of Akt/GSK-3beta/beta-catenin signaling pathway
is involved in survival of neurons after traumatic brain injury in
rats. Neurol Res. 34:400–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pérez-Álvarez MJ, del Maza MC, Anton M,
Ordoñez L and Wandosell F: Post-ischemic estradiol treatment
reduced glial response and triggers distinct cortical and
hippocampal signaling in a rat model of cerebral ischemia. J
Neuroinflammation. 9:1572012.
|
|
11
|
Mavila N, James D, Utley S, et al:
Fibroblast growth factor receptor-mediated activation of
AKT-β-catenin-CBP pathway regulates survival and proliferation of
murine hepatoblasts and hepatic tumor initiating stem cells. PloS
One. 7:e504012012.
|
|
12
|
Lei ZN, Liu F, Zhang LM, Huang YL and Sun
FY: Bcl-2 increases stroke-induced striatal neurogenesis in adult
brains by inhibiting BMP-4 function via activation of β-catenin
signaling. Neurochem Int. 61:34–42. 2012.PubMed/NCBI
|
|
13
|
Kingsbury Ma, Rehen SK, Contos JJ, Higgins
CM and Chun J: Non-proliferative effects of lysophosphatidic acid
enhance cortical growth and folding. Nat Neurosci. 6:1292–1299.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Valenta T, Hausmann G and Basler K: The
many faces and functions of β-catenin. EMBO J. 31:2714–2736.
2012.
|
|
15
|
Jin T, George Fantus I and Sun J: Wnt and
beyond Wnt: multiple mechanisms control the transcriptional
property of beta-catenin. Cell Signal. 20:1697–1704. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eves EM, Boise LH, Thompson CB, Wagner AJ,
Hay N and Rosner MR: Apoptosis induced by differentiation or serum
deprivation in an immortalized central nervous system neuronal cell
line. J Neurochem. 67:1908–1920. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rhee HJ, Nam JS, Sun Y, et al:
Lysophosphatidic acid stimulates cAMP accumulation and cAMP
response element-binding protein phosphorylation in immortalized
hippocampal progenitor cells. Neuroreport. 17:523–526. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang M, Zhong WW, Srivastava N, et al: G
protein-coupled lysophosphatidic acid receptors stimulate
proliferation of colon cancer cells through the β-catenin pathway.
Proc Natl Acad Sci USA. 102:6027–6032. 2005.PubMed/NCBI
|
|
19
|
Desbois-Mouthon C, Cadoret A, Blivet-Van
Eggelpoël MJ, et al: Insulin and IGF-1 stimulate the beta-catenin
pathway through two signalling cascades involving GSK-3beta
inhibition and Ras activation. Oncogene. 20:252–259. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Castellone MD, Teramoto H, Williams BO,
Druey KM and Gutkind JS: Prostaglandin E2 promotes colon cancer
cell growth through a Gs-axin-beta-catenin signaling axis. Science.
310:1504–1510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Verras M and Sun Z: Beta-catenin is
involved in insulin-like growth factor 1-mediated transactivation
of the androgen receptor. Mol Endocrinol. 19:391–398. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ortega F, Pérez-Sen R and Miras-Portugal
MT: Gi-coupled P2Y-ADP receptor mediates GSK-3 phosphorylation and
beta-catenin nuclear translocation in granule neurons. J Neurochem.
104:62–73. 2008.PubMed/NCBI
|
|
23
|
Yi F, Sun J, Lim GE, Fantus IG, Brubaker
PL and Jin T: Cross talk between the insulin and Wnt signaling
pathways: evidence from intestinal endocrine L cells.
Endocrinology. 149:2341–2351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iqbal S, Zhang S, Driss A, et al: PDGF
upregulates Mcl-1 through activation of β-catenin and
HIF-1α-dependent signaling in human prostate cancer cells. PloS
One. 7:e307642012.PubMed/NCBI
|
|
25
|
Hur EM and Zhou FQ: GSK3 signalling in
neural development. Nat Rev Neurosci. 11:539–551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu D and Pan W: GSK3: a multifaceted
kinase in Wnt signaling. Trends Biochem Sci. 35:161–168. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ng SS, Mahmoudi T, Danenberg E, et al:
Phosphatidylinositol 3-kinase signaling does not activate the wnt
cascade. J Biol Chem. 284:35308–35313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gherzi R, Trabucchi M, Ponassi M, et al:
The RNA-binding protein KSRP promotes decay of beta-catenin mRNA
and is inactivated by PI3K-AKT signaling. PLoS Biol. 5:e52006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takemaru K, Fischer V and Li FQ:
Fine-tuning of nuclear-catenin by Chibby and 14-3-3. Cell Cycle.
8:210–213. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luna-Ulloa LB, Hernández-Maqueda JG,
Santoyo-Ramos P, Castañeda-Patlán MC and Robles-Flores M: Protein
kinase C ζ is a positive modulator of canonical Wnt signaling
pathway in tumoral colon cell lines. Carcinogenesis. 32:1615–1624.
2011.
|
|
31
|
Paek H, Hwang JY, Zukin RS and Hébert JM:
β-Catenin-dependent FGF signaling sustains cell survival in the
anterior embryonic head by countering Smad4. Dev Cell. 20:689–699.
2011.
|
|
32
|
Holowacz T, Huelsken J, Dufort D and van
der Kooy D: Neural stem cells are increased after loss of
β-catenin, but neural progenitors undergo cell death. Eur J
Neurosci. 33:1366–1375. 2011.
|
|
33
|
Chen BY, Wang X, Wang ZY, Wang YZ, Chen LW
and Luo ZJ: Brain-derived neurotrophic factor stimulates
proliferation and differentiation of neural stem cells, possibly by
triggering the Wnt/β-catenin signaling pathway. J Neurosci Res.
91:30–41. 2013.PubMed/NCBI
|