Introduction
In previous years, with the rapid development of
molecular biology and tissue engineering, novel strategies on bone
repair from basic research to clinical applications have been
highly emphasized, in particular the role of the somatic stem cell.
At present, studies of stem cells focus largely on bone marrow
mesenchymal stem cells (BMSCs). In vitro and in vivo
studies have confirmed that BMSCs have the ability to differentiate
into osteoblasts, chondrocytes, adipocytes and neurons, as well as
numerous other cell types (1,2),
which indicates a wide application prospect in tissue engineering.
However, traditional bone marrow acquisition requires anesthesia
and often causes pain on puncture point, as well as harm to the
physical and psychological health of patients. The in vitro
purification rate of BMSCs is low at ~1×10 cells/30 ml bone marrow
(3). Numerous research groups have
devoted their studies to the identification of a novel type of stem
cell to replace BMSCs for bone repair. Skeletal muscle-derived stem
cells (MDSCs) are a type of pluripotent stem cell with multiple
differentiation potential and self-renewal ability. In addition,
these stem cells are abundant, easy to acquire and rapidly
amplified (4). The current study
aimed to investigate the isolation and culture of MDSCs, as well as
to verify their osteogenic potential in vitro. The results
are likely to provide a basis for future clinical applications to
promote bone healing in vivo.
Materials and methods
Isolation and culture of primary
cells
The medial muscles from the hind limbs of New
Zealand white rabbits were dissected (~5.0 g) under sterile
conditions (5). The study was
approved by the ethics committee of the First Affiliated Hospital
of Soochow University (Suzhou, China). Following careful removal of
the surrounding connective tissue, adipose tissue and capillaries,
the muscles were then sliced and ground into small sections using a
scalpel blade. The minced muscles were then thoroughly washed three
times with sterile phosphate-buffered saline (PBS) and digested
with 2 g/l type I collagenase (Invitrogen Life Technologies,
Carlsbad, CA, USA) for 1 h at 37ºC with 5% CO2 and 85%
humidity, as well as 2.5 g/l trypsin (Invitrogen Life Technologies)
for 40 min. Following digestion, the cell suspension was
centrifuged with an AllegraX-12R centrifuge (Beckman Coulter, Brea,
CA, USA) at 200 × g for 10 min. The cells were then filtered with
70- and 40-μm cell strainers to successively break down any
remaining cell clusters. Following further centrifugation (200 × g
for 10 min), the cell suspension was re-suspended in Dulbecco’s
modified Eagle’s medium (DMEM; Invitrogen Life Technologies)
containing 10% fetal bovine serum (FBS), 10% horse serum and 2%
penicillin/streptomycin, prior to being plated onto a dish. Each
dish was coded and cultured for 1 h in an incubator at 37ºC with 5%
CO2 and 85% humidity. Since the cells that adhered
rapidly within this one-hour incubation were predominantly
fibroblasts, the supernatant was withdrawn and gently transferred
to a new dish. This serial replating of the supernatant was
repeated ~5 times. In order to prevent cell differentiation,
cultures were maintained at semi-confluence and sub-cultured every
4–5 days with daily medium changes until adequate cell numbers were
obtained.
Identification of MDSCs
Cells were seeded in 12-well culture plates and
cultured in DMEM for 24 h. When the cells had stretched, they were
fixed with cold acetone for 5 min at −20ºC. Following rinsing with
PBS three times, the cells were incubated for 1 h at 37ºC.
Anti-desmin (Sigma, St Louis, MO, USA) and anti-myosin antibodies
(Invitrogen Life Technologies) were used as primary antibodies and
were diluted in PBS. A secondary antibody was applied for 1 h at
room temperature. Cells were rinsed with PBS and incubated at room
temperature with Hoechst 33342 fluorescent solution (Thermo Fisher
Scientific Pierce, Rockford, IL, USA) for 5 min, followed by
visualization using fluoroscopy (AXIOVERT 40C; Carl Zeiss AG,
Oberkochen, Germany). Positive cells were visualized in green,
while the nucleus was visualized in blue.
Cells (5×105) were re-suspended and
incubated at 4ºC for 30 min with 100 μl PBS containing 2 μl
anti-CD105-phycoerythrin antibody (Sigma-Aldrich). Following the
washing process, unconjugated primary antibodies were incubated
with a secondary goat anti-mouse IgG-FITC (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) for an additional 15 min at
4ºC. The cell suspension was then washed with PBS and labeled cells
were analyzed by flow cytometry (AllegraX-12R; Beckman Coulter).
Control samples were incubated with PBS instead of primary
antibody, followed by incubation with secondary antibody.
Osteogenic differentiation
MDSCs were seeded in 6-well plates at a density of
106 cells/well and cultured in osteogenic medium
containing DMEM with 20% FBS, 0.1 mM dexamethasone, 0.05 mM
ascorbic acid-2-phosphate, 10 mM β-glycerolphosphate, 100 U/ml
penicillin and 100 U/ml streptomycin. The cells cultured in basic
medium served as control and culture medium was changed every three
days.
Assessment of cell proliferation
Cell proliferation was quantitatively evaluated by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
assay. MDSCs were seeded in 96-well plates in medium containing 10%
FBS at a density of 5,000 cells/well. The MTT assay was performed
in triplicate. MTT (20 μl) was added to a final concentration of 5
mg/ml and the reaction mixture was incubated for 4 h at 37ºC. The
supernatant was removed and 150 μl DMSO was added to each well to
solubilize the water-insoluble purple formazan crystals. The
absorbency was measured at a wavelength of 590 nm.
Alkaline phosphatase (ALP) assay
MDSCs were seeded in 12-well plates and covered with
medium. Following 48 h, the medium was removed and ALP staining was
performed. All steps were performed in accordance with the
manufacturer’s instructions (Jiancheng Biotech, Guangzhou, China).
The staining was observed via microscope following four, eight, 12
and 16 days of differentiation. The ALP-positive cells stained
brown. For each experiment, a minimum of three plates were counted
and the experiments were repeated three times.
MDSCs were also seeded in 48-well plates and
incubated in medium. ALP activity was measured following four,
eight, 12 and 16 days of differentiation by the ALP assay kit
(BioVision, Inc., San Francisco, CA, USA). The cells were harvested
with lysis buffer. Total protein concentrations of supernatant were
determined by the Bradford method. An aliquot (10 μl) of
supernatant was added to 100 μl 50 mM p-nitrophenylphosphatase
hexahydrate containing 1 mM MgCl2 and the mixture was
incubated at 37ºC for 30 min. Absorption at 405 nm was measured
with a spectrophotometer. ALP activity per total protein (μg)
represented the millimoles of p-nitrophenol released following 30
min incubation at 37ºC.
Assay of mineralization
The mineralization of MDSCs was determined in
12-well plates using Alizarin red staining (ScienCell Research
Laboratories, Carlsbad, CA, USA) following four, eight, 12 and 16
days of differentiation. Following two PBS washes, the cells were
fixed with ice-cold 70% ethanol for 10 min and stained with 0.1%
Alizarin Red in Tris-HCl buffer solution (adjusted to pH 8.3) for
30 min at 37ºC. Following staining, MDSCs were rinsed three times
with distilled water followed by 70% ethanol.
Evaluation of osteogenic potential by
quantitative polymerase chain reaction (qPCR)
Osteogenic gene expression (osteocalcin and
osteopontin) was quantified by qPCR. Total RNA was extracted with
TRIzol reagent (Invitrogen Life Technologies) from MDSCs, according
to the manufacturer’s instructions. cDNA synthesis was performed
with 2 μg total RNA. qPCR was performed using the ABI Prism 7500
sequence-detection system (Applied Biosystems, Foster City, CA,
USA) with SYBR-Green, according to the manufacturer’s instructions.
For each sample of DNA, two test runs were performed; one used the
osteogenic gene primers and the other used the GAPDH gene primers
(Table I). Each running sample was
composed of 3 μl cDNA, 1 μl forward primer, 0.25 μl reverse primer
and 5 μl SYBR-green. Each cycle (40 cycles in total) included
denaturation at 50ºC, annealing at 95ºC and elongation at 60ºC.
Relative gene expression was normalized against GAPDH expression
and the data were presented as the fold change using the formula
2−ΔΔCT, as recommended by the manufacturer.
 | Table IPrimers used in quantitative
polymerase chain reaction. |
Table I
Primers used in quantitative
polymerase chain reaction.
| Gene | Primer sequence |
|---|
| Osteocalcin | Forward:
5′-CCGGGAGGAGATCTG;TGAAA-3′
Reverse: 5′-CTGCCTTCTTCCACAATTTTATCC-3′ |
| Osteopontin | Forward:
5′-GCCAGTTGCAGCCTTCTCA-3′
Reverse: 5′-GCCATGCCCAAGAGACAAAA-3′ |
| GAPDH | Forward:
5′-CGGACACGGACAGGATTGAC-3′
Reverse: 5′-CCAGACAAATCGCTCCACCA-3′ |
Results
Characterization of MDSCs
The number of adhered MDSCs was reduced within two
to three days. The cells assumed a spindle-shaped fibroblastic
appearance, proliferated rapidly and became confluent at one week
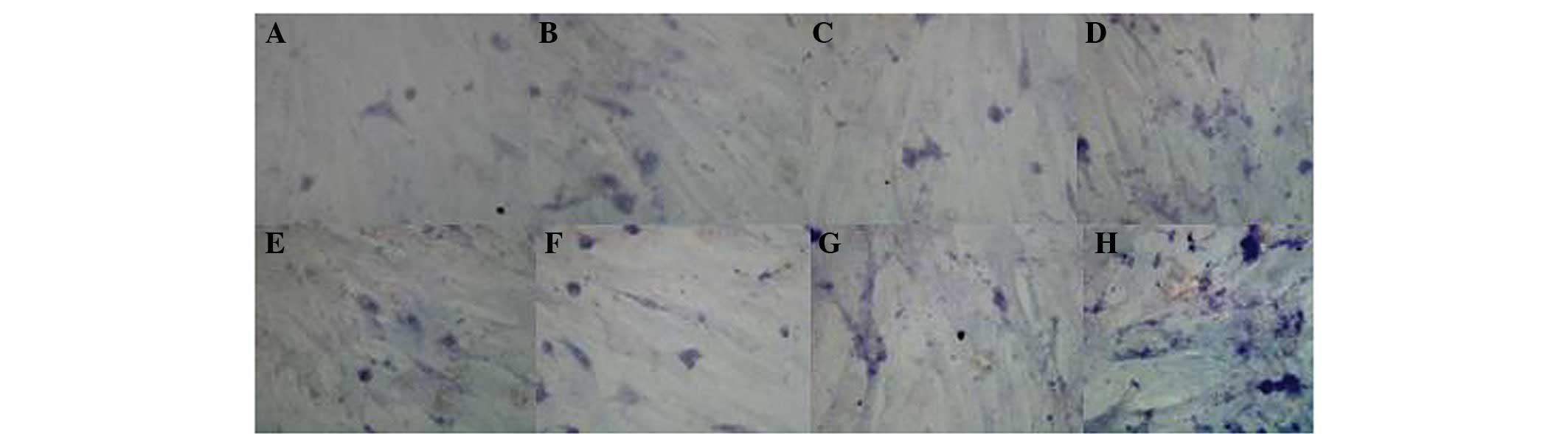
of culture. Immunohistochemical staining demonstrated that the
majority of detected cells expressed myogenic markers (desmin and
myosin; Fig. 1). Expression of
desmin is ubiquitous throughout the whole cell except for the
nucleus, while the expression of myosin is localized in the
nucleus. Flow cytometric analysis also demonstrated that the
majority of cells were positive for the MSC marker, CD105 (Fig. 2). When plated and cultured in
osteogenic medium, MDSCs changed from an elongated fibroblastic
appearance to a more rounded, cuboidal shape and formed an
extensive network of dense, multilayered nodules.
Assessment of cell proliferation
Based on the MTT assay, growth curves of
differentiated and undifferentiated MDSCs presented an ‘S’ shape.
The lag phase occurs at ~24 h, followed by the logarithmic phase of
growth and a growth-arrested phase. No significant difference was
observed on day four following osteogenic differentiation. However,
between days eight and 16, the cell proliferation rate of
differentiated MDSCs was significantly higher than that of
undifferentiated MDSCs (P<0.05). This growth curve shows
significantly higher cell growth rates in the differentiated MDSC
group than in the undifferentiated MDSC group (P<0.05; Fig. 3), indicating that the
differentiated MDSC population proliferated faster than the
undifferentiated MDSCs population.
ALP assay
The differentiated MDSCs stained positive for
membrane-bound ALP on day four and the staining became stronger at
day 16 (Fig. 4). Results indicated
that osteogenic differentiation stimulated the MDSCs to undergo
osteogenic differentiation, as judged by the production of ALP (an
early osteogenic marker) in response to osteogenic differentiation.
By contrast, undifferentiated MDSCs predominantly showed negative
reactions to ALP staining at any time point, demonstrating that
undifferentiated MDSCs did not undergo osteogenic differentiation.
Quantitation of ALP activity demonstrated that the ALP activity of
undifferentiated MDSCs was significantly lower than that of
corresponding differentiated MDSCs. ALP activities of
differentiated MDSCs increased between days four and 12. As time
progressed, the activity was higher, reaching a peak at day 16. By
comparison, the activities of undifferentiated MDSCs remained
stable at a low level (Fig.
5).
Mineralization assay
The well-defined mineralization nodules stained by
alizarin red staining were observed in differentiated MDSCs.
Between days four and 12, Alizarin red (+) nodules increased
constantly. At ~day 16, the progress of matrix mineralization
significantly step by step diffusely spread. However, no marked
mineralization nodules were found in undifferentiated MDSCs at any
time point (Fig. 6).
Analysis of the mRNA expression of
osteocalcin and osteopontin
Osteogenic differentiation had a clear positive
effect on the expression of two bone-associated proteins,
osteocalcin and osteopontin, in MDSCs and this expression was
markedly increased over time. The synthesis and secretion of
osteocalcin and osteopontin from differentiated MDSCs increased
gradually and attained a significantly higher level at day 16,
while the secretion of undifferentiated MDSCs was quite low and
stable at each time point (Fig.
7).
Discussion
Skeletal muscle has been extensively investigated as
a potential source for isolation of several stem cells over the
past few years (6). Satellite
cells located beneath the basal lamina of mature skeletal muscle
fibers, often referred to as ‘muscle stem cells’, have long been
considered monopotential stem cells capable of only giving rise to
cells of the myogenic lineage (7).
MDSCs, which may represent a predecessor of the satellite cell, are
considered to be distinct in that they may not be restricted to the
myogenic lineage or mesenchymal tissues and are able to
differentiate into multiple lineages (8).
Several studies have hypothesized that there are at
least two types of stem cells in skeletal muscle tissue; the
typical spindle muscle satellite cells and round MDSCs (9). Muscle satellite cells are myogenic
precursors, which are capable of regenerating skeletal muscle and
demonstrate self-renewal properties; however, they are considered
to be committed to the myogenic lineage (10). MDSCs are precursors of muscle
satellite cells and have a stronger capacity to proliferate and
broader potential of differentiation compared with muscle satellite
cells. These cells may differentiate into more specialized cells
with morphological and phenotypic features of cells including
neurocytes, hepatocytes, endotheliocytes, neurogliocyte, skeletal
muscle cells. MDSCs not only differentiate into mesodermal cells,
but also endodermal cells. By breaking the limit of the germ
layers, these cells exhibit wide application prospects (11,12).
Primary culture methods of MDSCs include the tissue
block method and digestion methods, of which the enzyme digestion
culture method is the most commonly used (8–10).
In the present study, mechanical shearing and enzyme digestion was
used to break the MDSC membrane and remove the connective tissues
interfering with cell growth. MDSCs dispersed into monocellular
suspension contribute to nutrient absorption and excretion of
metabolites from the medium. Since the mechanism of action and the
optimal condition of various types of digestive enzymes are not all
the same, stepwise digestion was performed using collagenase
combined with trypsin, which yielded 80–90% MDSCs. In addition,
MDSCs were purified by removing muscle fiber fragments and
nonmyogenic components in cell suspension liquid, in order to meet
the requirements for the next step. A suitable centrifugation
method (200 × g; 3 min) was used to precipitate the muscle fiber
fragments. If the centrifugal force is too large or the
centrifugation time is too long, MDSCs are likely to be damaged and
cell yield decreased. However, if the centrifugation time is too
short, nonmyogenic components mix with cells. In addition, cell
strainers of various sizes were also used to filter muscle fiber
fragments. For removal of the nonmyogenic components in mixed cell
suspension, the preplate technique was mainly utilized, which
isolates cell populations based on their differential adhesion
characteristics. The adherent ability of nonmyogenic components is
stronger than that of the MDSCs and the majority of early adherent
cells are nonmyogenic cells. Non-adherent cell suspensions were
removed and seeded in novel culture dishes and this preplate
procedure was repeated until MDSCs were pure. Regardless of certain
disadvantages, for example a time consuming (five to seven days for
adherence) and complex procedure, as well as a high possibility of
pollution, cell damage is minimal and cell purity may reach >80%
using this technique (13).
There is no generally established specific marker
for identification of multipotential stem cells from skeletal
muscle. Markers commonly used are expressed in various tissues and
with the prolongation of cell culture time, the cell behaviors and
surface markers are likely to have changed; thus rendering the
identification of MDSCs difficult. In the present study, a
combination of specific generally accepted molecular markers,
including stem cells and myogenic cells, was optimized to identify
MDSCs. CD105, a characteristic glycoprotein expressed on stem
cells, has frequently been used as a relatively specific marker for
the identification of mesenchymal stem cells. Desmin is a major
essential intermediate filament protein, as well as a specific
marker for muscle tissue and is used to distinguish between
myogenic and nonmyogenic tissues (14). The positive rate of desmin may
reach 90% in MDSCs, while in fibroblast and smooth muscle cells,
the rate of desmin-positive cells is only 15% (15). Myosin is the most common protein in
muscle cells and is a globulin responsible for the elastic and
contractile properties of muscle, combining with actin to form
actomyosin. It forms the thick filament of myofibrilla and is the
most abundant protein expressed in skeletal muscle, making up ~25%
of the total protein database (16). Based on this combination,
preliminary identification may be performed successfully.
MDSCs differentiated into multinucleated myotubes
and muscle fibers autonomously without any induction; in the
myoblast differentiation medium, they differentiated into myogenic
cells, including skeletal muscle cells, smooth muscle cells and
cardiocytes (17–20). Jackson et al(21) injected MDSCs to six lethally
irradiated mice. Following 12 weeks, all mice showed high-level
engraftment of muscle-derived cells representing all predominant
adult blood lineages. This indicated that cells derived from
skeletal muscle generate the major hematopoietic lineages. In the
present study, following culture with osteogenic differentiation
medium, MDSCs were positive for ALP and mineralized nodule
staining. Immunohistochemical staining and flow cytometric analysis
showed that desmin, myosin and CD105 were also positive in the
differentiated MDSCs, indicating that MDSCs had differentiated into
osteogenic cell lines. Although the mechanism of differentiation is
not yet clear, it may be that MDSCs directly differentiate into
osteogenic precursor cells or there were other stem cells in
skeletal muscle that differentiated into other osteoblasts. When
these precursor cells are mature, they may express various specific
markers of bone tissues. Following osteogenic differentiation, the
MDSCs in the present study gradually changed in morphology and
eventually formed round cells with a large body. In addition,
expression of ALP, osteocalcin and osteopontin was noted in these
cells. ALP is one of the early marker enzymes of maturity of
extracellular matrix and hydrolyzes organic phosphatase, increases
the local concentration of phosphoric acid, destroys inhibitors of
calcification and initiates the calcification process for promoting
mineralization of the extracellular matrix. ALP expression was
detected four days following osteogenic differentiation in the
present study and expression was significantly increased following
day 16. Osteopontin is a key factor of maturity of the
extracellular matrix and is associated with hydroxyapatite
deposition of extracellular matrix; it is important in
mineralization metabolism and reconstruction of bone. Osteocalcin
is a type of noncollagenous bone matrix protein secreted by mature
osteoblasts. It regulates the speed and direction of calcium
deposition, as a key factor of bone formation. In the present
study, the mRNA expression of these two proteins was less in
undifferentiated MDSCs; however, when the cells were cultured in
the osteogenic differentiation medium, expression was higher than
previously observed. This indicated that MDSCs had differentiated
into osteoblasts.
In the present study, MDSCs were successfully
isolated and cultured from rabbit skeletal muscle tissues, and
in vitro analyses indicated that the cells were capable of
differentiating into osteoblasts. Since the concept of cell therapy
has already been introduced into the clinical setting, MDSCs which
represent a valuable source of osteoprogenitors, exhibit the
potential for the repair of skeletal damage due to injury or
disease. Further studies are required in order to identify
optimization of cell isolation and expansion, as well as the
control of cell differentiation and transformation in various
environments.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81101370), Jiangsu
Science and Technology Support Program (Social Development) (grant
no. BE2011672) and Jiangsu Collegiate Natural Science Foundation
(grant no. 12KJB320008).
References
|
1
|
Strioga M, Viswanathan S, Darinskas A,
Slaby O and Michalek J: Same or not the same? Comparison of adipose
tissue-derived versus bone marrow-derived mesenchymal stem and
stromal cells. Stem Cells Dev. 21:2724–2752. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Osugi M, Katagiri W, Yoshimi R, Inukai T,
Hibi H and Ueda M: Conditioned media from mesenchymal stem cells
enhanced bone regeneration in rat calvarial bone defects. Tissue
Eng Part A. 18:1479–1489. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mauney JR, Kirker-Head C, Abrahamson L,
Gronowicz G, Volloch V and Kaplan DL: Matrix-mediated retention of
in vitro osteogenic differentiation potential and in vivo
bone-forming capacity by human adult bone marrow-derived
mesenchymal stem cells during ex vivo expansion. J Biomed Mater Res
A. 79:464–475. 2006. View Article : Google Scholar
|
|
4
|
Schindeler A, Liu R and Little DG: The
contribution of different cell lineages to bone repair: exploring a
role for muscle stem cells. Differentiation. 77:12–18. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Usas A, Ho AM, Cooper GM, Olshanski A,
Peng H and Huard J: Bone regeneration mediated by BMP4-expressing
muscle-derived stem cells is affected by delivery system. Tissue
Eng Part A. 15:285–293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shi H, Verma M, Zhang L, Dong C, Flavell
RA and Bennett AM: Improved regenerative myogenesis and muscular
dystrophy in mice lacking Mkp5. J Clin Invest. 123:2064–2077. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yan J, Gan L, Yang H and Sun C: The
proliferation and differentiation characteristics of co-cultured
porcine preadipocytes and muscle satellite cells in vitro. Mol Biol
Rep. 40:3197–3202. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bueno DF, Kerkis I, Costa AM, et al: New
source of muscle-derived stem cells with potential for alveolar
bone reconstruction in cleft lip and/or palate patients. Tissue Eng
Part A. 15:427–435. 2009.PubMed/NCBI
|
|
9
|
Asakura A: Stem cells in adult skeletal
muscle. Trends Cardiovasc Med. 13:123–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jankowski RJ, Deasy BM and Huard J:
Muscle-derived stem cells. Gene Ther. 1:642–647. 2002. View Article : Google Scholar
|
|
11
|
Wu X, Wang S, Chen B and An X:
Muscle-derived stem cells: isolation, characterization,
differentiation, and application in cell and gene therapy. Cell
Tissue Res. 340:549–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arsic N, Mamaeva D, Lamb NJ and Fernandez
A: Muscle-derived stem cells isolated as non-adherent population
give rise to cardiac, skeletal muscle and neural lineages. Exp Cell
Res. 314:1266–1280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huard J, Yokoyama T, Pruchnic R, Qu Z, Li
Y, Lee JY, Somogyi GT, de Groat WC and Chancellor MB:
Muscle-derived cell-mediated ex vivo gene therapy for urological
dysfunction. Gene Ther. 9:1617–1626. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cornelison DD and Wold BJ: Single-cell
analysis of regulatory gene expression in quiescent and activated
mouse skeletal muscle satellite cells. Dev Biol. 191:270–283. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei Y, Li Y, Chen C, Stoelzel K, Kaufmann
AM and Albers AE: Human skeletal muscle-derived stem cells retain
stem cell properties after expansion in myosphere culture. Exp Cell
Res. 317:1016–1027. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schiaffino S and Reggiani C: Myosin
isoforms in mammalian skeletal muscle. J Appl Physiol. 77:493–501.
1994.PubMed/NCBI
|
|
17
|
Lee JY, Cannon TW, Pruchnic R, Fraser MO,
Huard J and Chancellor MB: The effects of periurethral
muscle-derived stem cell injection on leak point pressure in a rat
model of stress urinary incontinence. Int Urogynecol J Pelvic Floor
Dysfunct. 14:31–37. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reinecke H, Poppa V and Murry CE: Skeletal
muscle stem cells do not transdifferentiate into cardiomyocytes
after cardiac grafting. J Mol Cell Cardiol. 34:241–249. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Herreros J, Prósper F, Perez A, Gavira JJ,
Garcia-Velloso MJ, Barba J, et al: Autologous intramyocardial
injection of cultured skeletal muscle-derived stem cells in
patients with non-acute myocardial infarction. Eur Heart J.
24:2012–2020. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Iijima Y, Nagai T, Mizukami M, Matsuura K,
Ogura T, Wada H, Toko H, Akazawa H, Takano H, Nakaya H and Komuro
I: Beating is necessary for transdifferentiation of skeletal
muscle-derived cells into cardiomyocytes. FASEB J. 17:1361–1363.
2003.PubMed/NCBI
|
|
21
|
Jackson KA, Mi T and Goodell MA:
Hematopoietic potential of stem cells isolated from murine skeletal
muscle. Proc Natl Acad Sci USA. 96:14482–14486. 1999. View Article : Google Scholar : PubMed/NCBI
|