Introduction
Osteosarcoma is one of the most common bone tumors,
particularly in teenagers. It is highly malignant with a poor
prognosis due to its high risk of relapse and metastasis (1). Conventional treatment includes
surgical resection combined with chemotherapy, which is determined
based on the scope of tumor invasion. Approximately 30% of patients
at early stages have distant metastasis following surgery and
chemotherapy (2). With advances in
molecular biology and cell biology in clinical oncology, a limited
understanding of the mechanisms underlying osteosarcoma at the
molecular level has been developed. The abnormal expression of
certain oncogenes (e-myc, e-ras and e-fos) and
anti-oncogenes (p53, Rb and pl6) has been found to be associated
with this disease. This pattern of gene expression may provide a
theoretical foundation of target gene selection for osteosarcoma
gene therapy (3).
Actin is the major cytoskeletal protein responsible
for maintaining cell shape and polarity, promoting cell migration
and cytokinesis by dynamic alterations in depolymerization and
repolymerization. Actin participates in various physiological and
pathological processes, including phagocytosis, endocytosis,
embryogenesis, organogenesis and angiogenesis (4). Recent studies have demonstrated that
actin reorganization is also involved in tumor cell migration,
invasion and metastasis (5–7).
Cofilin is a member of the actin depolymerization
factor (ADF)/cofilin family, which is able to reinforce actin
motion and assist cell movement and chemotaxis (8,9). The
LIM kinase 1 (LIMK1) pathway and the slingshot phosphatase pathway
are two effective and important pathways involved in regulating
cofilin activation. LIMK1 specifically phosphorylates cofilin and
thus regulates actin filament dynamics under the control of Rac,
while LIMK2 is important in regulating stress fiber and filopodia
formation and accompanying actin cytoskeletal dynamics,
specifically downstream of Rho and Cdc42. LIMK1 is a novel dual
specificity (serine/threonine and tyrosine) kinase that contains
two amino-terminal LIM domains (10). The LIMK1 gene is expressed
predominantly in the brain and in developing neural tissues
(11) and its deletion
(microdeletion of chromosome 7q11.23) is typically associated with
Williams syndrome (12). LIMK1 is
activated by phosphorylation at Thr-508 (in the kinase catalytic
domain) by ROCK and PAK. These activating kinases are downstream
kinases of the Rho family of small GTPases, Rho, Rac and Cdc42
(13–15). LIMK1 is also able to be
phosphorylated at Ser-323 (outside the catalytic domain) by
mitogen-activated protein kinase (MAPK)-activated protein kinase-2
(MAPKAPK2), which is a kinase downstream of p38 MAPK (16).
In the present study, we silenced the LIMK1 gene
using interferon RNA in the cultured human osteosarcoma cell lines
U2OS and MG63, and studied the effect and mechanisms of human
osteosarcoma cell proliferation following LIMK1 knockdown.
Materials and methods
Tissue samples and cell culture
The human osteosarcoma cell lines U2OS and MG63
(purchased from the Institute of Biochemistry and Cell Biology,
Shanghai Institutes for Biological Sciences, Chinese Academy of
Sciences, Shanghai, China) were cultured in high glucose Dulbecco’s
modified Eagle’s medium (H-DMEM; Gibco-BRL Carlsbad, CA, USA)
containing 10% fetal calf serum. Human fetal osteoblastic (hFOB)
1.19 cells (donated by the Pathology Laboratory of Jilin
University, Changchun, Jilin, China) were cultured in DMEM-F12
(Gibco-BRL) supplemented with 10% fetal calf serum. All three types
of cells were incubated at 37°C, saturated humidity and 5%
CO2.
Tumor samples from a total of 6 patients were
collected intraoperatively and stored at −80°C with written consent
obtained. The present study was approved by the Institutional
Review Board of Jilin University and informed consent was obtained
from the patients/patient’s families.
Immunohistochemical and hematoxylin and
eosin (HE) staining
Tumor tissue sections were incubated with 3%
H2O2 to inactivate endogenous peroxidase
followed by deparaffination and rehydration. The antigen was
restored with 3% proteinase K, followed by washing with
phosphate-buffered saline (PBS) and blocking with 10% goat serum
for 1 h at room temperature. The sections were then incubated for 1
h at room temperature with a mouse mAb targeting β-actin (1:500;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) or rabbit pAb
to LIMK1, p-LIMK1, cofilin or p-cofilin (1:100, Santa Cruz
Biotechnology, Inc.) to detect tumor cells. Biotinylated rabbit
anti-mouse immunoglobulin diluted 1:100 in 3% normal goat serum was
applied for 1 h and followed with peroxidase-conjugated horseradish
streptavidin-biotin complex for 10 min. Reaction sites were
visualized using diaminobenzidine as the chromogen and nuclei were
counterstained with hematoxylin. Paraffin-embedded sections were
dehydrated, mounted with neutral gum and observed under a light
microscope (Olympus, Tokyo, Japan).
Immunoprecipitation and western blot
analysis
Human osteosarcoma cells, U2OS and MG63, and human
osteoblast cells, HFOB1.19, were lysed with lysis buffer (150 mM of
NaCl, 1% Nonidet P-40, 50 mM of Tris-HCl, pH 8.0 and 20 μM of PMSF)
at 4°C for 30 min and centrifuged at 12,000 × g at 4°C for 15 min.
The supernatant was stored at −70°C. Equal amounts of protein (100
ng), 15 μl of protein A agarose beads and 1 μl of anti-LIMK1
antibody (Santa Cruz Biotechnology, Inc.) were added to the tubes
and rotated overnight at 4°C. The agarose-antibody-antigen
complexes were collected by centrifugation (20 sec at 12,000 × g).
The supernatant was carefully removed and the complexes were washed
twice with lysis buffer. The pellet was resuspended in gel-loading
buffer and the protein was denatured by boiling for 5 min. Protein
A agarose beads were removed by centrifugation at 12,000 × g for 20
sec and the supernatant was transferred to a fresh tube. The
proteins were separated by SDS-PAGE and analyzed by immunoblotting
analysis, as described previously (17), using primary antibodies against
LIMK1, P(T508)-LIMK1, cofilin and p-cofilin (1:500; Santa Cruz
Biotechnology, Inc.). Immunodetection was accomplished using a
HRP-conjugated goat anti-rabbit secondary antibody (1:1,000;
Bioworld Technology Co, Ltd., St. Louis Park, MN, USA) and protein
bands were developed with an ECL Chemiluminescent Substrate Reagent
kit (Invitrogen Life Technologies, Carlsbad, CA, USA) and analyzed
by Image J Software (National Institutes of Health, Bethesda, MD,
USA).
Knockdown of LIMK1
To assess the effect of LIMK1 knockdown on
insulin-induced cell proliferation, we generated an shRNA construct
using the pSUPER vector (Oligo Engine, Seattle, WA, USA), as
described previously (18,19). The 19-base targeting sequences used
in the present study were as follows: 5′-GCTGGAACAATGGCTAGAA-3′
(human LIMK1). As a control, we used a nontargeting sequence,
5′-TCTTCCCCCAAGAAAGATA-3′, which does not exist in the human
genome. MG63 cells were plated in 100 mm dishes (1.5×106
cells/dish) and cultured for 24 h followed by transfection with
pSUPER-LIMK1 or the control pSUPER vector. Transfected cells were
cultured for 24 h prior to being transferred into 96-well chamber
slides at 5×103 cells/well. The cells were continuously
cultured for 16 h followed by serum starvation for 4 h and then
cultured with or without 1 μg/ml of insulin for 24 h.
Cell proliferation assay
MG63 or transfected MG63 (pSUPER-LIMK1-MG63) cells
were seeded into 96-well plates. After achieving 70% confluence,
the cells were suspended in 100 μl of serum-free medium. After 24
h, insulin (1 μg/ml; Sigma, St. Louis, MO, USA) or 1,25-dihydroxy
vitamin D3 (1,25(OH)2D3; 10−7
mol/l; Sigma) was added into each well. The Cell Counting kit-8
(CCK-8, Dojindo Laboratories, Kumamoto, Japan) was used to assess
cell proliferation at 24, 48 and 72 h. To study the effect of
inhibitors, cells were incubated with 20 mM of PD98059, a selective
MEK1 inhibitor (Sigma), or 10 μM of LY294002, a potent inhibitor
for PI3K (Sigma). Cells were exposed to these inhibitors in
serum-free medium at 37°C for 1 h, followed by the addition of 1
μg/ml of insulin. The control group was treated with H-DMEM as a
standard solution. Cells in the culture plates were incubated at
37°C with 5% CO2 and 95% humidity. After 24 h, the
optical density of the wells was read at 450 nm. The CCK-8 (Dojindo
Laboratories) reagent (10 μl) was added into each well and
incubated at 37°C for 2 h and the optical density was read again at
450 nm. The cell proliferation was calculated according to the
manufacturer’s instructions.
Statistical analysis
All data are presented as the mean ± standard
deviation (mean ± SD). Statistical analysis was performed using
one-way ANOVA (including Newman-Keuls multiple comparison test) and
statistical analysis software Prism 4 (GraphPad Software, Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression of LIMK1, p-LIMK1, cofilin and
p-cofilin in human osteosarcoma cells
Human osteosarcomatous tissues were analyzed by HE
staining and intensity immunohistochemistry was performed using a
specific antibody to recognize the expression of LIMK1/p-LIMK1 and
cofilin/P-cofilin in human osteosarcoma tissues. We demonstrated
that tumor cells were distributed in a diffuse pattern or formed
cancer nests. The size and the degree of differentiation of the
cells varied. The cells demonstrated large heteromorphic
anachromasis and karyokinesis and a high nucleo-cytoplasmic ratio
(Fig. 1A and B). The expression of
cofilin/p-cofilin (Fig. 1C and D)
and LIMK1/p-LIMK1 (Fig. 1E and F)
in tumor parenchyma was clearly higher than that in the mesenchyme,
indicating that LIMK1 and cofilin were overexpressed in
osteosarcomatous parenchyma. This result was confirmed by western
blot analysis, which demonstrated that the expression levels of
LIMK1 in U2OS and MG63 cells were almost 2-fold of that in hFOB1.19
cells (Fig. 2). These data
indicated that the overexpression of LIMK1 may be associated with
osteosarcoma biological activity.
Effect of LIMK1 in insulin and
1,25(OH)2D3 mediated proliferation of human
osteosarcoma cells
Insulin exerts, via the insulin receptor α, a wide
range of biological responses affecting glucose, lipid and protein
metabolism as well as cell proliferation and survival (20,21).
The effect of insulin on the proliferation of MG63 cells was
assessed. The results demonstrated that insulin dose-dependently
promoted the proliferation of MG63, with an effective concentration
between 0.5 to 10 μg/ml (Fig. 3A).
MG63 cells were then treated with 1 μg/ml of insulin for 0, 24, 48
and 72 h (Fig. 3B). The results
demonstrated that the insulin-induced cell proliferation increased
with time and the effect of insulin was predominantly robust at 24
h.
We also studied the possible mechanisms responsible
for insulin-induced osteosarcoma cell proliferation. Osteosarcoma
cells were pretreated with the PI3K pathway inhibitor LY294002 (10
μM) or the MAPK pathway inhibitor PD98059 (20 μM) for 1 h prior to
24 h incubation with insulin. The results demonstrated that PD98059
had no impact on insulin-induced cell proliferation, whereas
LY294002 significantly inhibited insulin-induced cell proliferation
(Fig. 3C), indicating that
insulin-induced cell proliferation may be associated with the PI3K
pathway, but not the MAPK pathway.
LIMK1 activation leads to the phosphorylation and
deactivation of cofilin, which is a specific substrate of LIMK1
(22). Thus, we examined the
expression and activation of cofilin in human osteosarcoma cells by
western blotting. After MG63 cells were stimulated by insulin at
various concentrations for 24 h, the expression of p-cofilin and
p-LIMK1 was significantly increased compared with the control
group. Pretreatment of cells with LY294002 significantly inhibited
the insulin-induced increase of p-cofilin and p-LIMK1 expression
(Fig. 4). By contrast, cells
pre-treated with PD98059 presented no alterations (data not
shown).
Our data suggested that insulin-induced human
osteosarcoma cell proliferation was associated with the
LIMK1/cofilin signaling pathway, which is regulated by PI3K. To
determine the role of LIMK1 in this pathway, we constructed an
shRNA vector targeting LIMK1 (pSUPER-LIMK1). Western blotting
demonstrated that MG63 cells transfected with the LIMK1 shRNA
expressed little endogenous LIMK1 compared with cells transfected
with control shRNA (Fig. 5A),
indicating that the shRNA vector targeting LIMK1 (pSUPER-LIMK1) was
constructed successfully. Furthermore, the LIMK1 shRNA, but not the
control shRNA, blocked insulin-induced cofilin phosphorylation
(Fig. 5B). After the LIMK1 gene
was silenced by transfection of pSUPER-LIMK1, there was a
significant decrease in cell proliferation and insulin-induced
proliferation was completely inhibited (Fig. 5C), indicating that LIMK1 was
essential in regulating the basic and insulin-induced proliferation
of MG63 human osteosarcoma cells.
Additionally, we demonstrated that
1,25(OH)2D3, at a concentration of
10−7 mol/l, inhibited human osteosarcoma cell
proliferation in a time-dependent manner (Fig. 6A). Transfection of pSUPER-LIMK1
plasmids led to significant inhibition of osteosarcoma cell
proliferation. However, after 1,25(OH)2D3 was
added, no further inhibition was observed indicating that LIMK1 is
important in the inhibition of proliferation by
1,25(OH)2D3 (Fig. 6B). Western blot analysis
demonstrated that p-cofilin expression increased transiently at 24
h, followed by a decrease from 48 to 72 h following
1,25(OH)2D3 treatment (Fig. 6C). The activity of cofilin is
reversibly regulated by phosphorylation and dephosphorylation at
Ser-3, with the phosphorylated form being inactive. LIMK
phosphorylates cofilin at Ser-3 and thereby inhibits the actin
filament disrupting the activity of cofilin. The inactive
Ser-3-phosphorylated cofilin (p-cofilin) is dephosphorylated and
reactivated by slingshot (SSH) family protein phosphatases.
Previous studies have demonstrated that slingshot and LIMK
spatially and temporally regulate the expression of cofilin
(23,24). Thus, we hypothesized that the
inhibitory effect of 1,25(OH)2D3 on cell
proliferation may be associated with LIMK, slingshot or other
related proteins. However, further studies are needed to examine
this link.
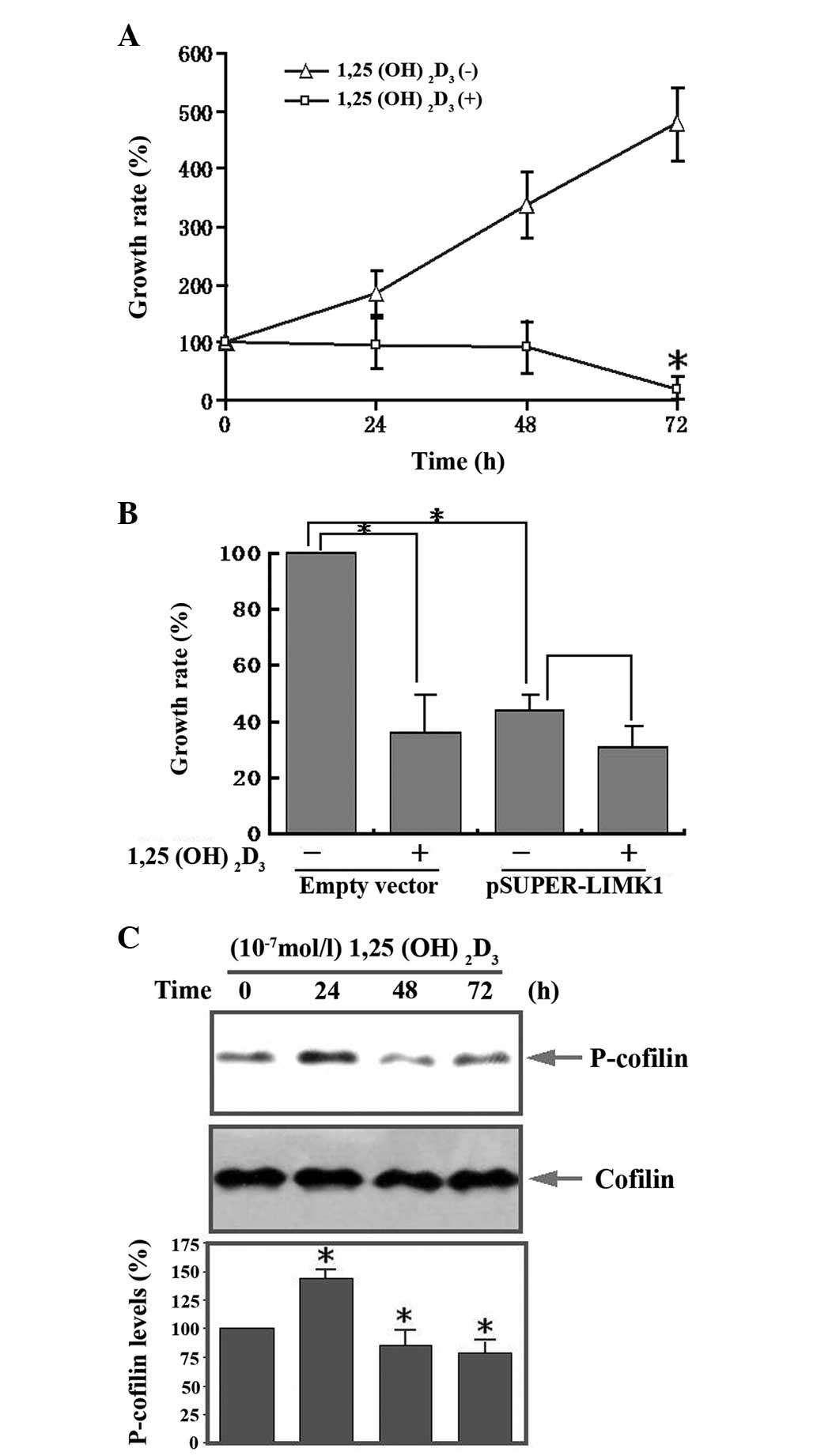 | Figure 6Effect of LIMK1 in
1,25(OH)2D3 mediated proliferation of human
osteosarcoma cells. (A) Serum-starved MG63 cells were stimulated
with 1,25(OH)2D3 (10−7 mol/l) for
the indicated times. Cell proliferation was detected with the CCK-8
kit, the OD values were measured at 450 nm. (B) MG63 cells were
transfected with LIMK1 shRNA and then stimulated with
1,25(OH)2D3. The control group was
transfected with an empty vector. Cell proliferation was detected
with the CCK-8 kit, the OD values were measured at 450 nm. (C)
Western blot analysis of the expression of p-cofilin at various
times following 1,25(OH)2D3 stimulation.
Serum-starved MG63 cells were stimulated with
1,25(OH)2D3 for the indicated times. Cell
lysates were immunoblotted with anti-p-cofilin and anti-cofilin
antibodies. The bottom panels show the relative p-cofilin levels.
LIMK1, LIM kinase 1; 1,25(OH)2D3,
1,25-dihydroxy vitamin D3; CCK-8, cell counting kit-8; OD, optical
density. |
Discussion
LIMK1, one of the LIMK proteins, is a
serine/threonine protein kinase that deactivates cofilin by
phosphorylation, thus participating in the rearrangement of the
actin cytoskeleton. Activation of LIMK1 is regulated by multiple
mechanisms and is important in extracellular stimulation and
cytoskeleton stability (25).
Recent studies have demonstrated that LIMK1 is overexpressed and
highly active in cells and tissues of certain malignant tumors,
including prostatic cancer and breast cancer (26), thus LIMK1 may be one of the key
molecules that stimulates tumor cell invasion and metastasis, or
possibly a new ‘oncogene’ (9). In
the present study, LIMK1 and p-LIMK1 expression in tumor parenchyma
cells are significantly higher than that in mesenchymal cells.
Furthermore, compared with human osteoblast cells HFOB 1.19,
expression of LIMK1 protein in human osteosarcoma cells U2OS and
MG63 was markedly higher, suggesting that overexpression of the
LIMK1 protein in human osteosarcoma cells may have a connection
with the biological characteristics of osteosarcoma.
Insulin is a hormone with various biological
effects, including the regulation of cell proliferation. Two major
pathways have been described in insulin signal transduction, one is
the PI3K pathway, the other is the MAPK pathway. In the present
study, insulin robustly enhanced MG63 cell proliferation in a time
and concentration-dependent manner. After cells were pretreated
with the PI3K pathway inhibitor LY294002, insulin-induced MG63 cell
proliferation was inhibited. By contrast, the MAPK pathway
inhibitor PD98059 had little effect, indicating that
insulin-induced MG63 cell proliferation primarily relies on the
PI3K pathway.
In the present study, we detected changes in cofilin
protein expression and activity in human osteosarcoma cells
stimulated by insulin at various concentrations for 24 h. Compared
with the control group, p-cofilin expression was significantly
increased, which indicated that insulin activates cofilin-related
signaling pathways. LIMK1 is important in cofilin activation, thus
cofilin phosphorylation, to a certain extent, is associated with
LIMK1 activation. However, insulin-induced activation of cofilin
was inhibited by LY294002, suggesting PI3K is involved in this
process.
We then transfected the pSUPER-LIMK1 plasmid into
MG63 cells to block transcription of the LIMK1 gene. Cell
proliferation was inhibited by blocking LIMK1 gene expression,
furthermore, insulin-induced cell proliferation was eradicated as
well. These data suggest that the expression of LIMK1 and the
activation of the insulin/LIMK1 pathway are crucial in regulating
MG63 cell proliferation.
The hormone variant of vitamin D3,
1,25(OH)2D3, regulates the expression of
genes through the interaction between the specificity receptor
vitamin D receptor (VDR), similarly to other steroid hormones. It
is also important in inhibiting tumor cell proliferation and
promoting tumor cell differentiation (27). Prior studies have demonstrated that
1,25(OH)2D3 inhibits the proliferation of
numerous tumors and synergistically promotes apoptotic death of
tumor cells in combination with other anticancer drugs (28,29).
However, the signaling transduction mechanisms by which
1,25(OH)2D3 inhibits tumor cell proliferation
are not clear. Using western blotting we demonstrated that
p-cofilin expression significantly increased following stimulation
with 10−7 mol/l of 1,25(OH)2D3 for
24 h. The proliferation of human osteosarcoma MG63 cells was
significantly inhibited after cells were stimulated with
10−7 mol/l of 1,25(OH)2D3. Cell
proliferation was also significantly inhibited after the plasmid
pSUPER-LIMK1 was transfected into human osteosarcoma cells,
however, there was no apparent further inhibition following the
addition of 1,25(OH)2D3, indicating that
LIMK1 and 1,25(OH)2D3 may share the same
pathway in inhibiting the proliferation of human osteosarcoma
cells.
In summary, LIMK1 and activation of the
insulin/PI3K/LIMK1 signaling pathway have major effects on human
osteosarcoma cell proliferation. The present study provides new
insight into LIMK1-regulated osteosarcoma genesis and
development.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81172000).
References
|
1
|
Picci P: Osteosarcoma (osteogenic
sarcoma). Orphamet J Rare Dis. 2:623–637. 2007.
|
|
2
|
Yamazaki D, Kurisu S and Takenawa T:
Regulation of cancer cell motility through actin reorganization.
Cancer Sci. 96:379–386. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gamberi G, Benassi MS, Bohling T, et al:
C-myc and c-fos in human osteosarcoma, prognostic value of mRNA and
protein expression. Oncology. 55:556–563. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Y, Shibasaki F and Mizuno K: Calcium
signal-induced cofilin dephosphorylation is mediated by Slingshot
via calcineurin. J Biol Chem. 280:12683–12689. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsai WC, Jin JS, Yu JC, et al: CD10,
actin, and vimentin expression in breast phyllodes tumors
correlates with tumor grades of the WHO grading system. Int J Surg
Pathol. 14:127–131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sebzda T, Saleh Y, Malicka-Blaszkiewicz M,
et al: Actin content and actin polymerization in hepatoma Morris
5123 tumor bearing rats after treatment with cysteine protease
inhibitor and vitamin E. J Exp Ther Oncol. 5:23–29. 2005.PubMed/NCBI
|
|
7
|
Amsellem V, Kryszke MH, Hervy M, et al:
The actin cytoskeleton-associated protein zyxin acts as a tumor
suppressor in Ewing tumor cells. Exp Cell Res. 304:443–456. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pavlov D, Muhlrad A, Cooper J, et al:
Actin filament severing by cofilin. J Mol Biol. 365:1350–1358.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang TY, DerMardirossian C and Bokoch GM:
Cofilin phosphatases and regulation of actin dynamics. Curr Opin
Cell Biol. 18:26–31. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Okano I, Hiraoka J, Otera H, et al:
Identification and characterization of a novel family of
serine/threonine kinases containing two N-terminal LIM motifs. J
Biol Chem. 270:31321–31330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bernard O, Geniatsus S, Kannourakis G and
Dringen R: Kiz-1, a protein with LIM zinc finger and kinase
domains, is expressed mainly in neurons. Cell Growth Differ.
5:1159–1171. 1994.PubMed/NCBI
|
|
12
|
Wouters CH, Meijers-Heijboer HJ, Eussen
BJ, et al: Deletions at chromosome regions 7q11.23 and 7q36 in a
patient with Williams syndrome. Am J Med Genet. 102:261–265. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Edwards DC, Sanders LC, Bokoch GM and Gill
GN: Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase
signalling to actin cytoskeletal dynamics. Nat Cell Biol.
1:253–259. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maekawa M, Ishizaki T, Boku S, et al:
Signaling from Rho to the actin cytoskeleton through protein
kinases ROCK and LIM-kinase. Science. 285:895–898. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ohashi K, Nagata K, Maekawa M, et al:
Rho-associated kinase ROCK activates LIM-kinase 1 by
phosphorylation at threonine 508 within the activation loop. J Biol
Chem. 275:3577–3582. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kobayashi M, Nishita M, Mishima T, Ohashi
K and Mizuno K: MAPKAPK-2-mediated LIM-kinase activation is
critical for VEGF-induced actin remodeling and cell migration. EMBO
J. 25:713–726. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okano I, Hiraoka J, Otera H, et al:
Identification and characterization of a novel family of
serine/threonine kinases containing two N-terminal LIM motifs. J
Biol Chem. 270:31321–31330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brummelkamp TR, Bernards R and Agami R: A
system for stable expression of short interfering RNAs in mammalian
cells. Science. 296:550–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takemura M, Mishima T, Wang Y, et al:
Ca2+/calmodulin-dependent protein kinase IV-mediated LIM kinase
activation is critical for calcium signal-induced neurite
outgrowth. J Biol Chem. 284:28554–28562. 2009.
|
|
20
|
Cheatham B and Kahn CR: Insulin action and
the insulin signaling network. Endocr Rev. 16:117–142.
1995.PubMed/NCBI
|
|
21
|
White MF: The insulin signalling system
and the IRS proteins. Diabetologia. 40:S2–S17. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wilson C, Vereshchagina N, Reynolds B, et
al: Extracellular and subcellular regulation of the PI3K/Akt
cassette: new mechanisms for controlling insulin and growth factor
signalling. Biochem Soc Trans. 35:219–221. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nishita M, Tomizawa C, Yamamoto M, et al:
Spatial and temporal regulation of cofilin activity by LIM kinase
and Slingshot is critical for directional cell migration. J Cell
Biol. 171:349–359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soosairajah J, Maiti S, Wiggan O, et al:
Interplay between components of a novel LIM kinase-slingshot
phosphatase complex regulates cofilin. EMBO J. 24:473–486. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takahashi H, Funakoshi H and Nakamura T:
LIM-kinase as a regulator of actin dynamics in spermatogenesis.
Cytogenet Genome Res. 103:290–298. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davila M, Frost AR, Grizzle WE and
Chakrabarti R: LIM kinase 1 is essential for the invasive growth of
prostate epithelial cells: implications in prostate cancer. J Biol
Chem. 278:36868–36875. 2003. View Article : Google Scholar
|
|
27
|
Makishima M and Honma Y: Ethacrynic acid
and 1 alpha, 25-dihydroxyvitamin D3 cooperatively inhibit
proliferation and induce differentiation of human myeloid leukemia
cells. Leuk Res. 20:781–789. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kawa S, Nikaido T, Aoki Y, et al: Vitamin
D analogues up-regulate p21 and p27 during growth inhibition of
pancreatic cancer cell lines. Br J Cancer. 76:884–889. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wilentz RE, Iacobuzio-Donahue CA, Argani
P, et al: Loss of expression of Dpc4 in pancreatic intraepithelial
neoplasia, evidence that DPC4 inactivation occurs late in
neoplastic progression. Cancer Res. 60:2002–2006. 2000.PubMed/NCBI
|



















