Introduction
Neuroblastoma (NB) is the most common type of
extracranial solid tumor of neuroectodermal cell origin in
children, occurring most frequently in the adrenal medulla and
sympathetic nervous system. Neuroblastoma accounts for 10% of
malignant solid tumors in children (1), with ~75% of cases occurring prior to
5 years of age. The biological characteristics of NB are diverse
and unique compared with other types of cancer of the nervous
system. In a number of infants, NB may spontaneously disappear. In
other infants, NB may differentiate into benign ganglioneuroma
following chemotherapy. However, in the majority of patients with
NB, metastases are observed at the time of diagnosis, with rapid
progression and mortality being a frequent outcome (2). There is a marked correlation between
amplification of the MYC family gene MYCN, rapid progression
of NB and poor prognosis (3). A
haploid genome, consisting of >10 copies of the MYCN
gene, is predictive of poor outcome independent of anatomic
staging, age and other clinical variables. By contrast, there is a
positive association between MYCN amplification and
malignant behavior in NB (4).
microRNAs (miRNAs) are endogenous non-coding RNAs of 22 nucleotides
that regulate the expression of proteins by binding to
complementary sequences in the 3′UTR of target genes. It is well
documented that miRNAs regulate a variety of biological processes,
including cell proliferation, apoptosis, differentiation and aging
(5,6). Accumulating evidence also indicates
that miRNAs are involved in a number of pathological conditions,
including cancer, cardiac infarction, arrhythmias, viral infection
and Alzheimer’s disease (7,8),
suggesting that miRNAs are a novel target for therapeutic
intervention. miRNA-202 (miR-202) functions as a promoter and
suppressor of tumor formation and a regulator of immune function
(9–12). The expression of miR-202 is high in
human endometrium and adipose tissue-derived stem cells, suggesting
that expression levels may be linked to cell cycle control.
Furthermore, alterations in miR-202 expression may be associated
with the formation of testicular tumors. In addition, miR-202 may
regulate the expression of the MYCN gene and thereby inhibit
the proliferation of neuroblastoma MNA Kelly cells, which generally
exhibit a high MCYN gene copy number (13). This study was designed to examine
whether MYCN is a target gene of miR-202 in the
neuroblastoma cell line LAN-5, whether miR-202 has a binding site
for the transcription factor E2F1 and to define a putative
regulatory pathway involving miR-202, MYCN and E2F1. A
dysfunctional E2F1/miR-202/MYCN signaling pathway may
contribute to malignant transformation or enhance the aggression of
neuroblastoma.
Materials and methods
Materials
LAN-5 cells were purchased from Tianjin Ke Ruijie
Biological Ltd.. (Tianjin, China), and HEK293T cells were purchased
from the Shanghai Institute of Biochemistry and Cell Biology
(Shanghai, China). RPMI-1640 medium and Lipofectamine 2000 were
purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
Reagents for quantitative polymerase chain reaction (qPCR) were
purchased from Takara Bio Inc. (Shiga, Japan). Antibodies against
E2F1 and MYCN were purchased from Sigma-Aldrich (St. Louis, MO,
USA). Genome sequencing was performed by the Beijing Huada Genomics
(Shenzhen, China). Primer premier 5.0 software was used for primer
design. The synthesis of primers and the PAGE purification process
was performed by Shanghai GenePharma Co., Ltd. (Shanghai, China).
The mature sequence of hsa-miR-202 (5′-AGAGGUAUAGGGCAUGGGAA-3′,
MIMAT0002811) and an miRNA inhibitor sequence were obtained
obtained from miRBase (http://www.mirbase.org/cgi-bin/get_seq.pl?acc=MIMAT0002811).
The negative control sequence and negative control inhibitor
sequence were purchased from Ruibo Biotechnology, Co., Ltd.
(Shanghai, China).
Cell transfection
The experimental culture groups included: i)
untransfected LAN-5 cells (control), ii) cells transfected with
miR-202 mimics (50 nmol/l), iii) cells transfected with the miR-202
scramble nucleotide sequence (negative control or NC, 50 nmol/l)
and iv) cells transfected with an miRNA inhibitor (NI; 100 nmol/l),
a negative control for NI (NCI; 50 nmol/l). Cells in log phase
growth were seeded on 6-well culture plates (2×105
cells/well) and transfected when the cell fusion rate reached 70%.
The RNA-Lipofectamine 2000 compound was added according to the
experimental group (1 μl/well to yield 20 nmol/l miRNA). After 36
h, the transfection medium was discarded, the cells were washed
with serum-free RPMI-1640 and cultured in RPMI-1640 supplemented
with 10% FBS. Fluorescence microscopy was used to determine
transfection efficiency.
Quantitative PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies) according to the manufacturer’s
instructions. Quantitative PCR analysis was performed using an
Applied Biosystems 7500 Real-Time PCR system (Foster City, CA,
USA). The expression level of 18S RNA was used as an internal
control for mRNAs and the U6 expression level was used as an
internal control for miRNAs. The primers used for quantitative PCR
analysis were as follows: U6 (forward, 5′-GCTTCGGCAGCACAT
ATACTAAAAT-3′ and reverse, 5′-CGCTTCACGAATTTGC GTGTCAT-3′), 18S
(forward, 5′-CCTGGATACCGCAGCT AGGA-3′ and reverse,
5′-GCGGCGCAATACGAATG CCCC-3′); miR-202 (RT primer,
5′-CTCAACTGGTGTCGTG GAGTCGGCAATTCAGTTGAGTTCCCAT-3′; forward,
5′-ACACTCCAGCTGGGAGAGGTATAGGGCATGG-3′ and reverse,
5′-CTCAACTGGTGTCGTGGA-3′) and MYCN (forward,
5′-CCTGAGCGATTCAGATGA-3′ and reverse, 5′-CATAGTTGTGCTGCTGGT-3′).
The expression level was calculated using the 2−ΔΔCt
method.
Western blot analysis
LAN-5 cells were lysed in RIPA buffer containing a
proteinase inhibitor cocktail (Biocolor BioScience &
Technology, Shanghai, China). The protein concentration was
determined using bicinchoninic acid (Bioss, Beijing, China).
Protein was separated on 10% SDS-PAGE at 30 μg/gel lane and
electrotransferred to nitrocellulose membranes. Membranes were
incubated with a primary antibody against MYCN (rabbit polyclonal;
Cell Biotech, Tianjin, China) at 4°C overnight, washed extensively
with 0.1% Tween-20 in PBS and incubated with a secondary antibody
conjugated to horseradish peroxidase (1:1,000; Pharmingen, Becton
Dickinson, San Diego, CA, USA) at room temperature for 3 h.
Immunolabeling was visualized using the ECL system (Amersham, UK).
Expression levels were normalized to the gel loading control and
expressed as fold changes compared with baseline (pretransfection)
values.
Luciferase reporter assay
The 3′UTR fragments of the MYCN gene were
obtained by PCR amplification and cloned separately into multiple
cloning sites of the psi-CHECK™-2 luciferase miRNA expression
reporter vector. HEK293T cells were transfected with miR-202 mimic,
miR-202 inhibitor, a control miRNA, a miRNA inhibitor or empty
plasmid using Lipofectamine 2000 (Invitrogen Life Technologies)
according to the manufacturer’s instructions. Nucleotide
substitution mutation analysis was performed using direct oligomer
synthesis of MYCN 3′UTR sequences. The constructs were
verified by sequencing. Luciferase activity was measured using the
dual luciferase reporter assay system kit (Promega, Madison, WI,
USA) according to the manufacturer’s instructions on a Tecan M200
luminescence reader.
Rapid amplification of 5′cDNA ends
(5′RACE)
The 5′RACE measurements were conducted according to
the manufacturer’s instructions (Invitrogen Life Technologies).
Briefly, total RNA was extracted from LAN-5 cells with TRIzol
reagent (Invitrogen Life Technologies). PCR reactions were
performed using the universal sense primer provided in the 5′RACE
kit and antisense primers (miR-202 outer, 5′-TTAGGCCAG
ATCCTCAAAGAAG-3′, miR-202 inter, ATAGGAAAAAG GAACGGCGG) specific
for the miR-202 coding sequence. The PCR product was cloned and
sequenced.
Chromatin immunoprecipitation (ChIP)
assay
Neuroblastoma cells were cultured in 10-cm dishes at
5×106/dish. Ice-cold PBS (10 ml) was added to each dish,
followed by panning and washing on a horizontal shaker (3×1 min).
Formaldehyde (1% in PBS) was used for crosslinking covalently
stabilized protein-DNA complexes. The ultrasonic slicing method was
used to cut DNA fragments bound to protein into 200–1,000 bp
fragments. Following centrifugation, the supernatant was collected,
added to Protein A Agarose and mixed for 1 h. A 10 μl aliquot was
reserved as the input for later use. Antibodies against E2F1 were
added to the remaining supernatant for co-immunoprecipitation and
incubated with Protein A Agarose. Following precipitation with low
salt solution and centrifugation, the supernatant was discarded and
the protein-DNA pellet separated by elution to obtain the DNA
template. The gene-specific primers were designed according to the
sequence of miR-202 (5′-GTTCTGCTGCTGCCGAGCGAG-3′ and
5′-CCTGGCTCAGCACTCTTCTCACA-3′). Quantitative PCR was performed to
measure target DNA levels in the purified DNA products.
Data analysis
The results are the averages of at least three
independent experiments from separately treated and transfected
cultures. Data are expressed as the mean ± SD. Statistical
comparisons were made by one-way analysis of variance (ANOVA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Expression of miR-202 in LAN-5
transfection groups
After the cells were transfected with the miR-202
mimic, miR-202 inhibitor or appropriated controls for 36 h, qPCR
was performed to assess miR-202 expression (Fig. 1). Expression was significantly
higher in cultures transfected with miR-202 mimic compared with
untransfected controls, while expression was significantly
inhibited (P<0.05) in the group transfected with the inhibitor
(NI group) compared with untransfected cultures or cultures
transfected with the inhibitor control transcript (NIC group).
 | Figure 1The expression of miR-202 after
transfection with miR-202 mimics and miR-202 inhibitor in LAN-5
cells. *P<0.05 vs. control and NI, NC, NCI groups.
Bars: 1, control group; 2, miR-202 mimics group; 3, NI group; 4, NC
group; 5, NCI group. NC, negative control; NI, mRNA inhibitor; NCI,
negative control for NI; miR-202, microRNA-202. |
Effect of miR-202 on expression of
MYCN
After neuroblastoma cells in each group were
transfected with miR-202 mimic, miR-202 inhibitor or left
untransfected for 48 h, qPCR was performed to detect the relative
expression levels of MYCN mRNA. There was no significant difference
in MTCN mRNA expression among the miR-202 mimic group, blank
control group (untransfected) and the miRNA negative control group
(P>0.05; Fig. 2A). However,
western blot analysis of the MYCN protein revealed significant
differences among the miR-202 mimic group and the negative control
group (P<0.05) (Fig. 2B and C),
suggesting that miR-202 inhibits translation of MYCN.
 | Figure 2The effects of miR-202 mimics on the
expression of MYCN. (A) The MYCN mRNA expression
levels were determined by quantitative PCR, *P>0.05
vs. control and NC group. (B) The MYCN protein expression
levels were determined by western blotting, *P<0.05
vs. control and NC group. (C) Western blot electrophoresis diagram.
Lanes: 1, control group; 2, negative group; and 3, miR-202 mimics
group. PCR, polymerase chain reaction; NC, negative control; NI,
mRNA inhibitor; NCI, negative control for NI; miR-202,
microRNA-202. |
miR-202 binds to the MYCN 3′UTR
TargetScan (human 6.2 version), a miRNA target gene
prediction software application, predicted two miR-202 binding
sites at 505 and 869 bp in the MYCN 3′UTR sequence. The
full-length sequence of the MYCN 3′UTR (910 bp) was cloned
downstream of the luciferase gene in the psiCHECK carrier to
construct the psiCHECK-2-MYCN 3′UTR carrier. LAN-5 cells
were cotransfected with the miR-202 mimic vector and vectors
carrying mutations in the MYCN 3′UTR-binding site 1 (505
bp), binding site 2 (869 bp) or both, to confirm direct binding of
miR-202 and MYCN 3′UTR. Cotransfection of LAN-5 cells with
miR-202 mimic and psiCHECK-2 MYCN 3′UTR significantly
inhibited luciferase activity (P<0.05), while transfection with
vectors carrying a mutation at binding sites 1 and 2 or both
vectors had little effect on luciferase activity, indicating that
miR-202 may directly regulate the expression of MYCN by
binding to target sites within the MYCN 3′UTR sequence
(Fig. 3).
 | Figure 3The interaction between miR-202 and
MYCN was detected by the sensor reporter. (A) Comparison of
luciferase activity of plasmid-transfected cloned MYCN-3′UTR
*P>0.05 vs. control and NC. (B) The luciferase
activity comparison of plasmid-transfected cloned
mut-MYCN-3′UTR, ΔP>0.05 vs. control and NC.
(C) The target sequence for miR-202 at MYCN 3′-UTR. Bars: 1,
control; 2, miR-202 mimic; 3, NC; 4, NI and 5, NCI groups. NC,
negative control; NI, mRNA inhibitor; NCI, negative control for NI;
miR-202, microRNA-202. |
Transcription initiation site for miR-202
determined by 5′RACE assay
Downstream-specific primers were designed to amplify
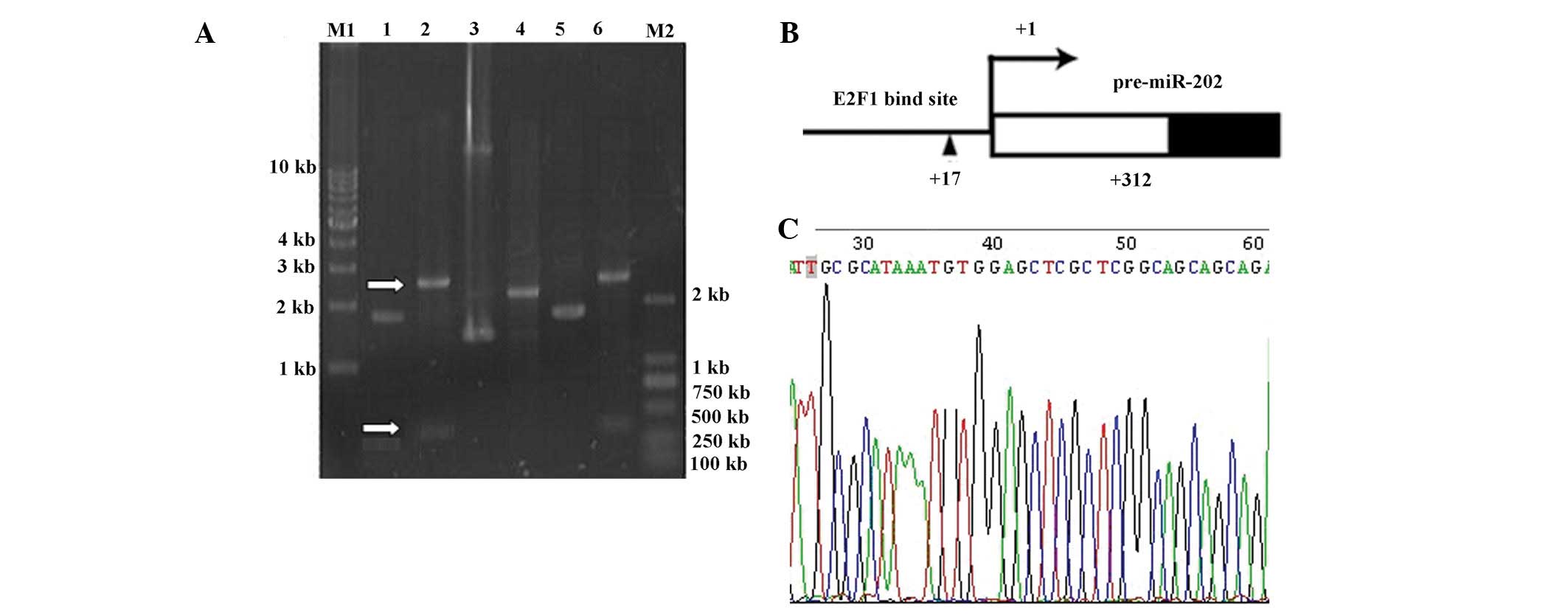
the miR-202 gene by PCR (Fig. 4).
Specific bands were observed corresponding to miR-202-1, -2 and -3.
The miR-202-1 band was the largest at >300 bp. The primary
transcriptional copy of miR-202 was ~300 bp. The DNA fragments were
separated from the protein, recovered from the gel and cloned on
the T-vector for sequencing. Sequences of the acquired DNA
fragments were compared and the start site for transcription was
determined to be a T-vector 312 bp upstream of the miR-202
stem-loop sequence. ConSite, a transcription factor prediction
software (obtained at http://asp.ii.uib.no:8090/cgi-bin/CONSITE/consite)
predicted an E2F1 binding site +17 bp upstream of the initiation
site of miR-202.
Binding of E2F1 to the miR-202 promoter
as revealed by ChIP assay
ChIP assay was used to verify the predicted binding
sites of E2F1 +17 bp upstream of the miR-202 initiation site.
First, western blot analysis was used to detect the expression of
E2F1 in LAN-5 cells. Electrophoresis of ChIP assay produced
positive bands for the E2F1 antibody group but none for the
negative control IgG group or blank group (water). Results from
quantitative PCR showed that in lysates, including the E2F1
antibody, the relative expression of E2F1 amplified by the
E2F1ChIPF/R primer was ~18-fold higher than that in the negative
group, suggesting that the DNA was isolated using the E2F1 antibody
containing the predicted miR-202 promoter sequence. These results
indicate that E2F1 may directly bind to the promoter sequence of
miR-202 (Fig. 5).
Discussion
Amplification and overexpression of the MYCN
oncogene is frequently observed in NB. Tumors with the MYCN
amplification exhibit an invasive growth pattern, malignant
proliferation of cancer cells, a rapid increase in tumor volume,
rupture through the tumor capsule and invasion of surrounding
tissues, early systemic metastasis, poor prognosis and high
mortality (14). This study of the
molecular mechanisms of MYCN regulation by miR-202 defines a
potential regulatory pathway for control of cell proliferation and
a promising therapeutic target for the treatment of NB (15).
miRNAs regulate a variety of critical biological
processes by controlling the expression of target genes, including
genes involved in cell proliferation, apoptosis and malignant
transformation (16). miRNAs may
inhibit translation or activate mRNA degradation through
complementary pairing with a 3′UTR sequence (17). Bioinformatics prediction provides a
convenient and powerful method for identifying potential miRNA
target genes (18). TargetScan,
the standard software for prediction of miRNA target genes,
predicted two possible binding sites on the MYCN 3′UTR for
miR-202 and it was confirmed that they are indeed regulatory sites
for MYCN expression. miR-202 overexpression significantly reduced
the luciferase activity of a vector containing MYCN 3′UTR
and mutation of these potential miR-202 binding sites significantly
reduced the inhibitory effect of miR-202 on luciferase activity
(Fig. 3). Through these direct
binding sites for miR-202 on MYCN, miR-202 overexpression
may significantly reduce the expression of MYCN protein (Fig. 2B and C). However, miR-202 had no
effect on the expression of MYCN mRNA (Fig. 2A), indicating that miR-202 binding
to the 3′UTR suppresses MYCN expression at the post-transcriptional
level.
The E2F1 transcription factor is the most
significant activator governing the transition from G1 to S phase
and thus, is a key regulator of cell proliferation. The Rb/E2F
complex is a critical intermediary step in this process (19). In normal cells, Rb in the low
phosphorylation state and E2F, form a complex that inhibits the
transcription of the proto-oncogenes c-myc, c-fos and others to
inhibit cell growth. By contrast, highly phosphorylated Rb and E2F
initiates downstream gene expression and promotes cell cycle
progression (20,21). Thus, E2F plays a dual role
depending on the phosphorylation status of Rb. Previous studies
have shown that E2F activity is required for MYCN
overexpression in neuroblastoma cells (13,14).
In IMR-32 cells, overexpression of cP16INK4A may shift Rb to the
low phosphorylation state, decreasing E2F1 activity and
downregulating MYCN expression (22–24).
In the current study, E2F1 was observed to bind to the miR-202
promoter and miR-202 was observed to directly target MYCN
(Figs. 4 and 5). Previous studies demonstrated that
miR-202 directly binds to MYCN in SMS-KMN cells, thereby
downregulating MYCN expression. A potential negative
feed-forward loop may exist among E2F1, miR-202 and MYCN.
Under normal circumstances, E2F1 indirectly inhibits MYCN
activity by upregulating miR-202, thus preventing excessive
activation of MYCN by E2F1. A dynamic balance among these
three molecules may maintain normal cell growth. However, this
balance is disturbed in nascent neuroblastoma cells, with eventual
loss of MYCN regulation and malignant transformation.
Therefore, elucidation of the molecular mechanisms
regulating E2F1, miR-202 and MYCN may identify novel
molecular targets for the treatment of NB and aid in the
development of novel therapeutic strategies.
Acknowledgements
This study was supported by grants from the
Guangdong Science and Technology Department Social Development
Projects (no. 2011B080702011) and the Technical New Star of
Zhujiang, Pan Yu districts, Guangzhou.
References
|
1
|
Brodeur GM: Neuroblastoma: biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Attiyeh EF, London WB, Mossé YP, Wang Q,
Winter C, et al: Chromosome 1p and 11q deletions and outcome in
neuroblastoma. N Engl J Med. 353:2243–2253. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brodeur GM, Seeger RC, Schwab M, Varmus HE
and Bishop JM: Amplification of N-myc in untreated human
neuroblastomas correlates with advanced disease stage. Science.
224:1121–1124. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar
|
|
5
|
Lim LP, Lau NC, Weinstein EG, et al: The
microRNAs of Caenorhabditis elegans. Genes Dev. 17:991–1008.
2003.
|
|
6
|
Berezikov E, Guryev V, van de Belt J, et
al: Phylogenetic shadowing and computational identification of
human microRNA genes. Cell. 120:21–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cai B, Pan Z and Lu Y: The roles of
microRNAs in heart diseases: a novel important regulator. Curr Med
Chem. 17:407–411. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Liang Y and Lu Q: MicroRNA
epigenetic alterations: predicting biomarkers and therapeutic
targets in human diseases. Clin Genet. 74:307–315. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Couzin J: MicroRNAs make big impression in
disease after disease. Science. 319:1782–1784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hawkins SM, Creighton CJ, Han DY, et al:
Functional microRNA involved in endometriosis. Mol Endocrinol.
25:821–832. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Novotny GW, Nielsen JE, Sonne SB, et al:
Analysis of gene expression in normal and neoplastic human testis:
new roles of RNA. Int J Androl. 30:316–326. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cho JA, Park H, Lim EH and Lee KW:
MicroRNA expression profiling in neurogenesis of adipose
tissue-derived stem cells. J Genet. 90:81–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Buechner J, Tømte E, Haug BH, et al:
Tumour-suppressor microRNAs let-7 and mir-101 target the
proto-oncogene MYCN and inhibit cell proliferation in
MYCN-amplified neuroblastoma. Br J Cancer. 105:296–303. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei JS, Song YK, Durinck S, Chen QR, Cheuk
AT, et al: The MYCN oncogene is a direct target of miR-34a.
Oncogene. 27:5204–5213. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schulte JH, Horn S, Otto T, Samans B,
Heukamp LC, et al: MYCN regulates oncogenic MicroRNAs in
neuroblastoma. Int J Cancer. 122:699–704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–333. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hornstein E and Shomron N: Canalization of
development by microRNAs. Nat Genet. 38(Suppl): S20–S24. 2006.
View Article : Google Scholar
|
|
18
|
Peterson KJ, Dietrich MR and McPeek MA:
MicroRNAs and metazoan macroevolution: insights into canalization,
complexity, and the Cambrian explosion. Bioessays. 31:736–747.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harbour JW and Dean DC: The Rb/E2F
pathway: expanding roles and emerging paradigms. Genes Dev.
14:2393–2409. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Trimarchi JM and Lees JA: Sibling rivalry
in the E2F family. Nat Rev Mol Cell Biol. 3:11–20. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rabinovich A, Jin VX, Rabinovich R, Xu X
and Farnham PJ: E2F in vivo binding specificity: comparison of
consensus versus nonconsensus binding sites. Genome Res.
18:1763–1777. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen CR, Kang Y, Siegel P and Massagué J:
E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to
c-myc repression. Cell. 110:19–32. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lacerte A, Korah J, Roy M, Yang XJ, Lemay
S and Lebrun JJ: Transforming growth factor-beta inhibits
telomerase through SMAD3 and E2F transcription factors. Cell
Signal. 20:50–59. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Emmrich S and Pützer BM: Checks and
balances: E2F-microRNA crosstalk in cancer control. Cell Cycle.
9:2555–2567. 2010. View Article : Google Scholar : PubMed/NCBI
|