Introduction
Gastric cancer is the fourth most common malignancy
and the second leading cause of cancer mortality worldwide
(1). Numerous studies indicate
that the epithelial growth factor receptor (EGFR) is overexpressed
in various cancer tissues, including gastric cancer, and the
majority of human epithelial cancers are marked by a high activity
of the EGFR (2–5). Numerous studies have demonstrated
that the activation of the EGFR is able to initiate several signal
transduction cascades, which lead to the stimulation of cell
proliferation, migration, invasion, metastasis, angiogenesis and
the inhibition of apoptosis (6,7). Due
to its close association with the origin, development, metastasis
and invasion of cancer cells, the EGFR was the first growth factor
receptor to be proposed as a target for cancer therapy (8,9).
Anti-EGFR therapies have been intensively developed for the
treatment of multiple cancer types and EGFR inhibitors are
currently under investigation.
Type II cGMP-dependent protein kinases (PKG II) is a
serine-threonine (ser-thr) kinase. It has been identified for
>30 years, however certain new functions of this kinase are just
being discovered, including its important role in regulating cell
proliferation and apoptosis (10–12).
Recently, accumulating data demonstrate that PKG II is a potential
cancer inhibitor. For example, it was demonstrated that PKG II was
able to inhibit the proliferation of human neuroglioma cells and
colonic cancer cells (13,14). Data from our laboratory
demonstrated that PKG II inhibited the proliferation and migration
of gastric cancer cell lines (15,16).
Furthermore, we demonstrated that PKG II was able to inhibit EGF
initiated signal transduction of the MAPK-mediated pathway by
inhibiting the EGF-induced activation of the EGFR in gastric and
breast cancer (17,18). Since the mammalian ligands of EGFR
include transforming growth factor-α (TGF-α), heparin-binding
EGF-like growth factor, amphiregulin, betacellulin (BTC),
epiregulin (EPR) and epigen besides EGF (19), whether PKG II has an inhibitory
effect on the activation of EGFR induced by these ligands merits
further investigation. The present study was designed to elucidate
whether PKG II was able to inhibit the activation of the EGFR
induced by EGF, TGF-α, BTC and EPR.
Materials and methods
Cell lines and reagents
The human gastric cancer cell line AGS was purchased
from ATCC (Manassas, VA, USA; ATCC® number:
CRL-1793TM). Adenoviral vectors encoding β-galactosidase
(Ad-LacZ) and PKG II (Ad-PKG II) were provided by Dr Gerry Boss and
Dr Renate Pilz at the University of California (San Diego, CA,
USA). Dulbecco’s Modified Eagle’s Medium (DMEM) and new-born calf
serum (NBCS) were purchased from Gibco-BRL (Grand Island, NY, USA).
The antibody against PKG II was purchased from Abgent Biotechnology
(San Diego, CA, USA). Rabbit anti-EGFR antibody was purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Mouse
anti-p-EGFR (tyr1068), rabbit anti-p-ERK1/2 (thr 202/tyr 204) and
rabbit anti-ERK1/2 antibodies were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The horseradish peroxidase
(HRP)-conjugated secondary antibodies were obtained from Jackson
ImmunoResearch Laboratories (West Grove, PA, USA). The cellular
permeable cGMP analog 8-pCPT-cGMP was acquired from Calbiochem (San
Diego, CA, USA). Recombinant human EGF, TGF-α, EPR and BTC were
obtained from PeproTech (Rocky Hill, NJ, USA).
Electrochemiluminescence (ECL) reagents were purchased from
Millipore (Billerica, MA, USA). All other reagents used were of
analytical grade.
Preparation of the adenoviral
vectors
293A cells were transfected with an adenoviral
vector encoding LacZ and PKG II, respectively and cultured for up
to 10 days until a cytopathic effect was apparent. The cells and
the culture medium were harvested and underwent three freeze-thaw
cycles. The supernatant containing adenoviruses (Ad-LacZ and Ad-PKG
II) were used to infect new 293A cells to amplify adenoviruses. The
amplified adenoviral preparations were titrated and the pfu/ml was
determined and stored at −80°C until use.
Cell culture and infection with the
adenoviral vectors
AGS cells were cultured in DMEM supplied with 10%
NBCS and maintained at 37°C in a humidified incubator with 95% air
and 5% CO2. The medium was changed every 2 days and the
cells were sub-cultured at confluence. On the day prior to
infection, cells were freshly planted at 70–80% confluence and
infection was performed with a multiplicity of infection of
100%.
Western blotting
Proteins extracted from whole cells were separated
by 10% SDS-PAGE and were transferred onto the PVDF membranes. The
primary antibodies were incubated overnight at 4°C and the
corresponding secondary antibodies were incubated for 1 h at room
temperature, with three washes following each incubation. ECL
reagents were used to demonstrate the positive bands on the
membrane.
Results
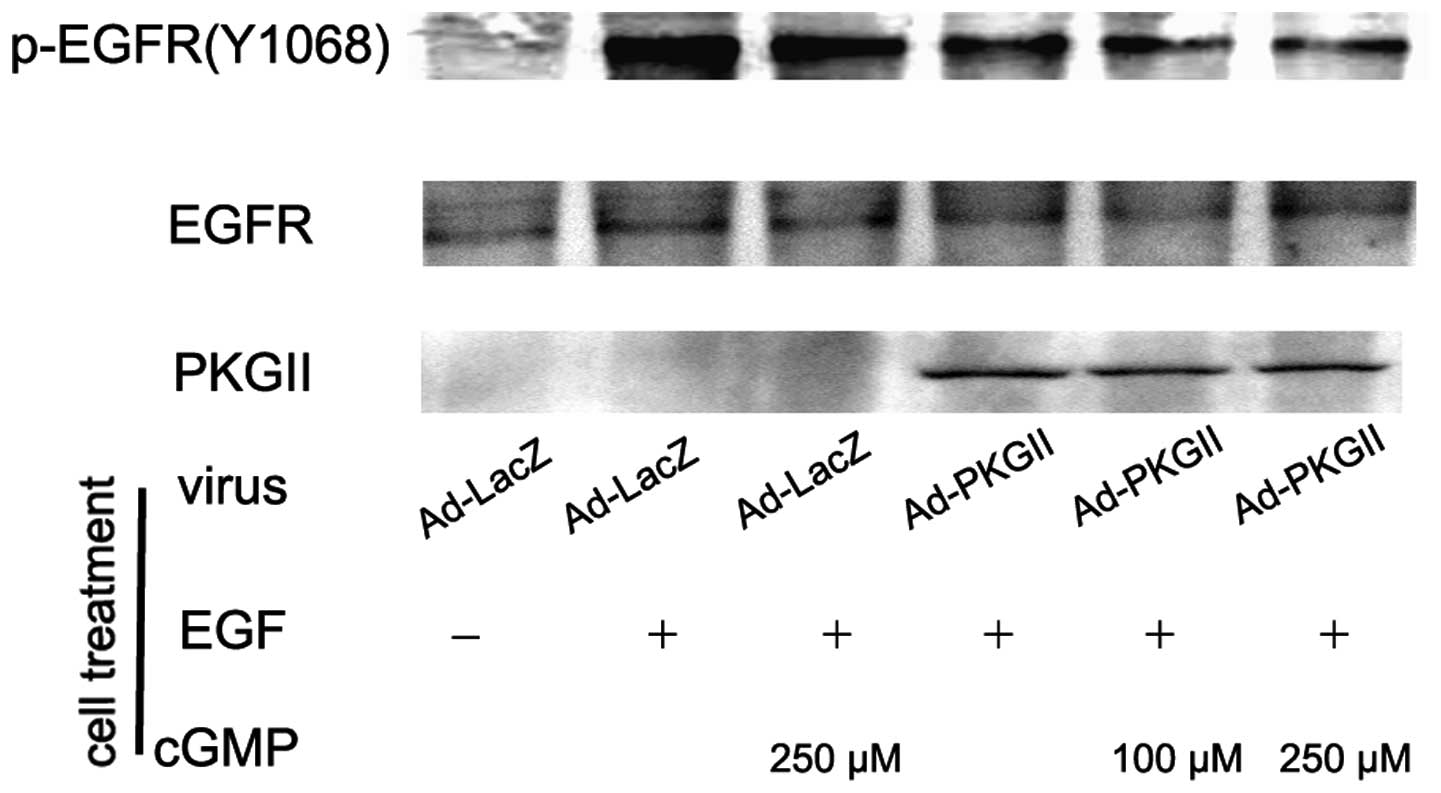
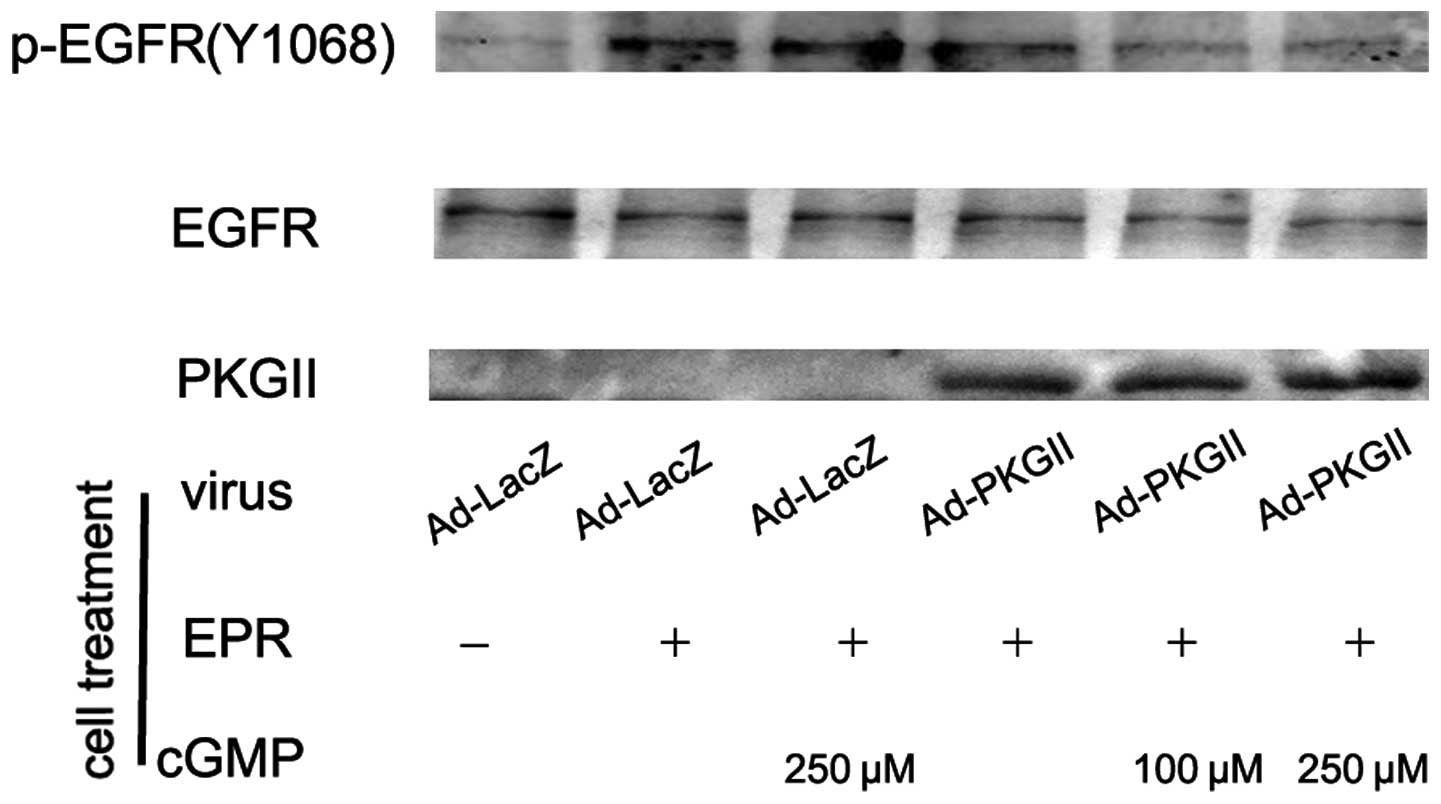
PKG II inhibits EGF, TGF-α, BTC and EPR
induced tyr 1068 phosphorylation of the EGFR
When ligands bind with EGFR, the binding between the
ligand and the EGFR causes auto-phosphorylation of the receptor.
There are several auto-phosphorylation sites which are connected to
different signal transduction pathways. Tyr 1068 is one of the
auto-phosphorylation sites of the EGFR and is associated with
MAPK/ERK-mediated signaling. In this experiment, we investigated
the inhibitory effect of PKG II on the tyr 1068 phosphorylation of
the EGFR in differently treated AGS cells by using western
blotting. The results demonstrated that the infection with Ad-PKG
II caused a clear increase in PKG II expression (Ad-PKG II groups).
The treatments with EGF (100 ng/ml, 5 min), TGF-α (100 ng/ml, 5
min), BTC (100 ng/ml, 5 min) and EPR (100 ng/ml, 5 min) caused
clear increases in tyr 1068 phosphorylation of the EGFR,
respectively. In cells infected with Ad-PKG II and stimulated with
8-pCPT-cGMP, the phosphorylation caused by the growth factors was
significantly decreased (Fig.
1–4). This indicated that PKG
II was able to inhibit EGF, TGF-α, BTC and EPR-induced tyr 1068
phosphorylation/activation of the EGFR.
PKG II inhibits the ERK activation
induced by EGF, TGF-α, BTC and EPR
Activation of the EGFR is able to initiate several
signal transduction pathways. Among them, the MAPK/ERK-mediated
pathway is the one closely associated with the proliferation and
differentiation of cells. MAPK/ERK is the key component of this
pathway and phosphorylation at thr 202 and tyr 204 residues of ERK1
and thr 185 and tyr 187 residues of ERK2 is required for full
enzymatic activation. In this experiment, the antibody against
p-ERK1/2 (thr 202/tyr 204) was applied in western blotting to
detect the dual phosphorylation of ERK. The results demonstrated
that within 5 min after adding EGF (100 ng/ml), TGF-α (100 ng/ml),
BTC (100 ng/ml) and EPR (100 ng/ml) to cell culture medium,
phosphorylation of ERK1/2 in AGS cells significantly increased and
the phosphorylation was inhibited by pre-infecting the cells with
Ad-PKG II and activating the enzyme with 8-pCPT-cGMP (Fig. 5).
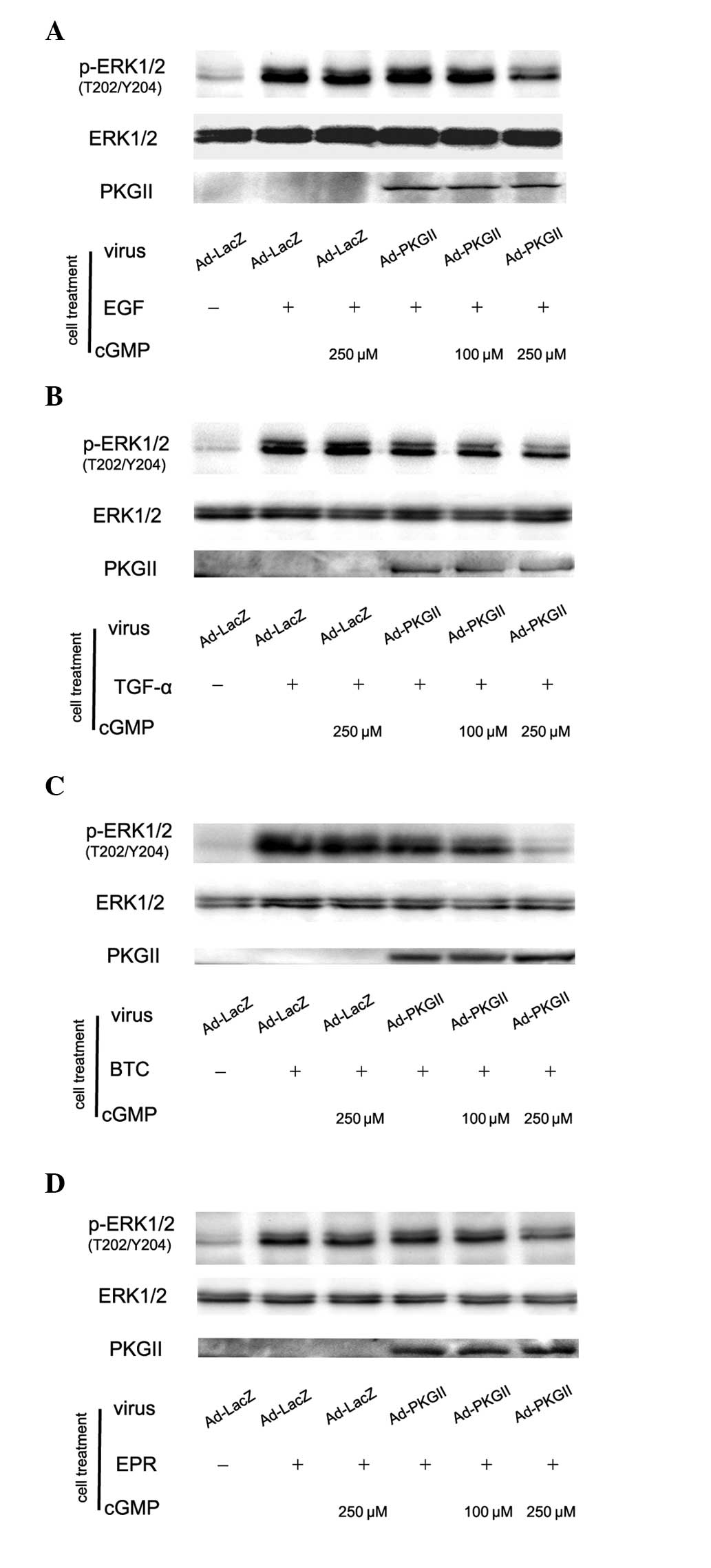 | Figure 5PKG II inhibits the phosphorylation of
ERK induced by EGF, TGF-α, BTC and EPR, respectively. AGS cells
were infected with Ad-PKG II for 48 h, serum starved overnight,
incubated with 8-pCPT-cGMP for 1 h and then stimulated with EGF
(100 ng/ml), TGF-α (100 ng/ml), BTC (100 ng/ml) and EPR (100
ng/ml), respectively for 5 min. Western blotting was applied to
detect the phosphorylation of ERK. Within 5 min after adding the
ligands to culture medium, phosphorylation of ERK significantly
increased. (A) Ad-LacZ + EGF group; (B) Ad-LacZ + TGF-α group; (C)
Ad-LacZ + BTC group; (D) Ad-LacZ + EPR group. The phosphorylation
was inhibited by pre-infecting the cells with Ad-PKG II and
stimulating the enzyme with 8-pCPT-cGMP. (A) Ad-PKG II + cGMP + EGF
groups; (B) Ad-PKG II + cGMP + TGF-α groups; (C) Ad-PKG II + cGMP +
BTC groups; (D) Ad-PKG II + cGMP + EPR groups. The results shown
are representative of three separate experiments. PKG II, type II
cGMP-dependent protein kinase; ERK, extracellular signal-regulated
kinase; EGF, epidermal growth factor; TGF-α, transforming growth
factor-α, BTC, betacellulin; EPR, epiregulin; Ad-LacZ, adenoviral
vectors encoding β-galactosidase; Ad-PKG II, adenoviral vectors
encoding PKG II; cGMP, cyclic guanosine monophosphate. |
Discussion
The EGFR belongs to the ErbB receptor tyrosine
kinase family, which includes erbB1 (EGFR), erbB2 (HER2), erbB3
(HER3) and erbB4 (HER4). EGFR is a 170 kDa protein that consists of
an extracellular domain, a transmembrane domain and a cytoplasmic
domain (20). The activation of
EGFR depends on its binding with ligands and the consequent
dimerization and auto-phosphorylation of the receptor. When the
EGFR is activated, it is able to recruit effector proteins to its
phosphorylated C-terminal sub-domain and initiate the effector
protein-mediated signaling pathways. Increased expression and
activation of the EGFR and its ligands are causally associated with
the progression of gastric cancer and poor prognosis (21,22).
The ligands of the EGFR include EGF, TGF-α, BTC and
EPR. They are structurally and functionally related proteins and
are able to bind and activate the EGFR. These ligands and the
consequent EGFR signaling are important in regulating the
proliferation, survival, migration and differentiation of various
cancer cells (19).
EGF is thought to be involved in neoplastic
formation and the progression of gastric cancer. EGF is a single
polypeptide built of 53 amino acids and is important in
intercellular interactions. Co-operation between the EGF of one
cell and the receptor (EGFR) on the adjacent cell leads to certain
biological effects, including migration, growth or morphological
changes in the cell (23).
Normally, EGF stimulates the growth or exerts a trophic effect in
numerous tissues and is important in the proliferation,
differentiation and maturation of embryonic cells. However, EGF and
the EGFR, apart from the key role they play in physiological
development, also take part in neoplastic transformation. Increased
expression of EGF and the EGFR have been observed in various
malignancies, including gastric carcinoma (24).
TGF-α is produced in macrophages, brain cells and
keratinocytes, and induces epithelial development. It shares only
~30% structural homology with EGF, however it is also able to bind
to the EGF receptor with similar affinity and signal via the EGFR
(25). TGF-α is upregulated in
certain types of human cancer. Several studies demonstrated that
overexpression of growth factors in the gastric mucosa was
implicated in the pathogenesis of gastric cancer (26) and gastric cancer cells grew in
response to EGF/TGF-α activation of the EGFR in an autocrine loop
(27).
BTC was originally identified in conditioned media
from a pancreatic β-cell tumor line (28,29).
There is evidence confirming that BTC is a potent mitogen for
vascular smooth muscle cells and retinal pigment epithelial cells
(28,30). BTC has also been detected in a
variety of tumor-derived cell lines and tumors in situ
(30,31).
EPR was initially isolated from the conditioned
medium of mouse tumorigenic fibroblasts NIH3T3/clone T7 cells
(32). There are extremely low
expression levels of EPR transcripts in the majority of normal
tissues except in the peripheral blood monocytes and the uterus
(33,34), however several cancer cells and
cytokine-induced smooth muscle cells overexpress EPR mRNA (33). It is known that EPR is a more
potent mitogen than EGF for several types of normal cells (32,35)
and is important in the development and progression of epithelial
malignancies in an autocrine/paracrine manner (36).
For a long time, PKG II has been implicated in
several physiological functions, including intestinal secretion,
bone growth and learning and memory. However, certain new functions
of PKG II are being discovered recently, including the
participation of PKG II in mechanotransduction in osteoblasts and
the role of PKG II in the regulation of sodium channels (37,38).
More importantly, accumulating data indicated that PKG II may be a
potential cancer suppressor because it is critical in the
proliferation, migration and apoptosis of cancer cells (15,16,18).
Our previous results demonstrated that PKG II was able to inhibit
EGF-induced tyr 1068 phosphorylation of the EGFR in gastric cancer
and breast cancer cells (16–18),
raising the question of whether PKG II is able to inhibit tyr 1068
phosphorylation of the EGFR induced by other ligands. Results in
the present study not only demonstrated that EGF, TGF-α, BTC and
EPR were able to induce tyr 1068 phosphorylation of EGFR and thr
202 and tyr 204 phosphorylation of ERK, but also confirmed that PKG
II was able to inhibit these phosphorylations. This revealed that
PKG II has a wide-ranging inhibitory effect on the activation of
the EGFR induced by diverse ligands and adds further evidence to
support PKG II as a potential cancer suppressor.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81272755, no. 31040002, no.
81201959 and no. 31100974) and the Innovation Grant of Jiangsu
University. We thank Dr Gerry Boss and Dr Renate Pilz, University
of California (San Diego, CA, USA) for providing the adenoviral
constructs.
References
|
1
|
Shah MA and Schwartz GK: Treatment of
metastatic esophagus and gastric cancer. Semin Oncol. 31:574–587.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Becker JC, Muller-Tidow C, Serve H, et al:
Role of receptor tyrosine kinases in gastric cancer: new targets
for a selective therapy. World J Gastroenterol. 12:3297–3305.
2006.PubMed/NCBI
|
|
3
|
Arnold D, Peinert S, Voigt W, et al:
Epidermal growth factor receptor tyrosine kinase inhibitors:
present and future role in gastrointestinal cancer treatment: a
review. Oncologist. 11:602–611. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johnston JB, Navaratnam S, Pitz MW, et al:
Targeting the EGFR pathway for cancer therapy. Curr Med Chem.
13:3483–3492. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Astsaturov I, Cohen RB and Harari PM:
EGFR-targeting monoclonal antibodies in head and neck cancer. Curr
Cancer Drug Targets. 6:691–710. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oda K, Matsuoka Y, Funahashi A and Kitano
H: A comprehensive pathway map of epidermal growth factor receptor
signaling. Mol Syst Biol. 1:2005.0010. 2005.PubMed/NCBI
|
|
7
|
Yarden Y and Shilo BZ: SnapShot: EGFR
signaling pathway. Cell. 131:10182007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arteaga CL: Overview of epidermal growth
factor receptor biology and its role as therapeutic taget in human
neoplasia. Semin Oncol. 29:3–9. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Krozely P: Epidermal growth factor
receptor tyrosine kinase inhibitors: evolving role in the treatment
of solid tumors. Clin J Oncol Nurs. 8:163–168. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cook AL and Haynes JM: Protein kinase G
II-mediated proliferative effects in human cultured prostatic
stromal cells. Cell Signal. 16:253–261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Swartling FJ, Ferletta M, Kastemar M, et
al: Cyclic GMP-dependent protein kinase II inhibits cell
proliferation, Sox9 expression and Akt phosphorylation in human
glioma cell lines. Oncogene. 28:3121–3131. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fallahian F, Karami-Tehrani F, Salami S,
et al: Cyclic GMP induced apoptosis via protein kinase G in
oestrogen receptor-positive and -negative breast cancer cell lines.
FEBS J. 278:3360–3369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Swartling FJ, Ferletta M, Kastemar M, et
al: Cyclic GMP-dependent protein kinase II inhibits cell
proliferation, Sox9 expression and Akt phosphorylation in human
glioma cell lines. Oncogene. 28:3121–3131. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang R, Kwon IK, Thangaraju M, et al: Type
2 cGMP-dependent protein kinase regulates proliferation and
differentiation in the colonic mucosa. Am J Physiol Gastrointest
Liver Physiol. 303:G209–G219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen YC, Ren F, Sang JR, et al: Type II
cGMP-dependent protein kinase inhibits proliferation of the gastric
cancer cell line BGC-823. Mol Med Rep. 3:361–366. 2010.PubMed/NCBI
|
|
16
|
Jiang L, Lan T, Chen Y, et al: PKG II
inhibits EGF/EGFR-induced migration of gastric cancer cells. PLoS
One. 8:e616742013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu Y, Chen Y, Qu R, et al: Type II
cGMP-dependent protein kinase inhibits EGF-triggered signal
transduction of the MAPK/ERK-mediated pathway in gastric cancer
cells. Oncol Rep. 27:553–558. 2012.PubMed/NCBI
|
|
18
|
Lan T, Chen Y, Sang J, et al: Type II
cGMP-dependent protein kinase inhibits EGF-induced MAPK/JNK signal
transduction in breast cancer cells. Oncol Rep. 27:2039–2044.
2012.PubMed/NCBI
|
|
19
|
Harris RC, Chung E and Coffey RJ: EGF
receptor ligands. Exp Cell Res. 284:2–13. 2003. View Article : Google Scholar
|
|
20
|
Prigent SA and Lemoine NR: The type I
(EGFR-related) family of growth factor receptors and their ligands.
Prog Growth Factor Res. 4:1–24. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barnard JA, Beauchamp RD, Russell WE, et
al: Epidermal growth factor-related peptides and their relevance to
gastrointestinal pathophysiology. Gastroenterology. 108:564–580.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
D’Agnano I, D’Angelo C, Savarese A, et al:
DNA ploidy, proliferative index, and epidermal growth factor
receptor: expression and prognosis in patients with gastric
cancers. Lab Invest. 72:432–438. 1995.PubMed/NCBI
|
|
23
|
Wells A: EGF receptor. Int J Biochem Cell
Biol. 31:637–643. 1999. View Article : Google Scholar
|
|
24
|
Takemura S, Yashiro M, Sunami T, et al:
Novel models for human scirrhous gastric carcinoma in vivo. Cancer
Sci. 95:893–900. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang C, Lv X, Jiang C, et al: Transforming
growth factor alpha (TGFα) regulates granulosa cell tumor (GCT)
cell proliferation and migration through activation of multiple
pathways. PLoS One. 7:e482992012.
|
|
26
|
Dias A, Garcia C, Majewski M, et al:
Gastric juice prostaglandins and peptide growth factors as
potential markers of chronic atrophic gastritis, intestinal
metaplasia and gastric cancer: their potential clinical
implications based on this pilot study. Dig Dis Sci. 56:3220–3225.
2011. View Article : Google Scholar
|
|
27
|
Yoshida K, Kyo E, Tsujino T, et al:
Expression of epidermal growth factor, transforming growth
factor-alpha and their receptor genes in human gastric carcinomas;
implication for autocrine growth. Jpn J Cancer Res. 81:43–51. 1990.
View Article : Google Scholar
|
|
28
|
Sasada R, Ono Y, Taniyama Y, et al:
Cloning and expression of cDNA encoding human betacellulin, a new
member of the EGF family. Biochem Biophys Res Commun.
190:1173–1179. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dunbar AJ and Goddard C:
Structure-function and biological role of betacellulin. Int J
Biochem Cell Biol. 32:805–815. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shing Y, Christofori G, Hanahan D, et al:
Betacellulin: a mitogen from pancreatic beta cell tumors. Science.
259:1604–1607. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seno M, Tada H, Kosaka M, et al: Human
betacellulin, a member of the EGF family dominantly expressed in
pancreas and small intestine, is fully active in a monomeric form.
Growth Factors. 13:181–191. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Toyoda H, Komurasaki T, Uchida D, et al:
Epiregulin. A novel epidermal growth factor with mitogenic activity
for rat primary hepatocytes. J Biol Chem. 270:7495–7500.
1995.PubMed/NCBI
|
|
33
|
Toyoda H, Komurasaki T, Uchida D, et al:
Distribution of mRNA for human epiregulin, a differentially
expressed member of the epidermal growth factor family. Biochem J.
326:69–75. 1997.PubMed/NCBI
|
|
34
|
Das SK, Das N, Wang J, et al: Expression
of betacellulin and epiregulin genes in the mouse uterus temporally
by the blastocyst solely at the site of its apposition is
coincident with the “window” of implantation. Dev Biol.
190:178–190. 1997.PubMed/NCBI
|
|
35
|
Komurasaki T, Toyoda H, Uchida D, et al:
Mechanism of growth promoting activity of epiregulin in primary
cultures of rat hepatocytes. Growth Factors. 20:61–69. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shirakata Y, Komurasaki T, Toyoda H, et
al: Epiregulin, a novel member of the epidermal growth factor
family, is an autocrine growth factor in normal human
keratinocytes. J Biol Chem. 275:5748–5753. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rangaswami H, Marathe N, Zhuang S, et al:
Type II cGMP-dependent protein kinase mediates osteoblast
mechanotransduction. J Biol Chem. 284:14796–14808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nie HG, Chen L, Han DY, et al: Regulation
of epithelial sodium channels by cGMP/PKGII. J Physiol.
587:2663–2676. 2009. View Article : Google Scholar : PubMed/NCBI
|