Introduction
Bone development and homeostasis is achieved by a
strict balance between bone formation by osteoblasts and resorption
by osteoclasts. Defective bone resorption results in osteopetrosis,
an inherited disorder encompassing a clinical and genetical
heterogenous group of conditions. Three main forms are classified
on the basis of inheritance patterns, age of onset and severity.
These are the autosomal recessive severe (ARO; MIM 259700),
intermediate autosomal (IAO) and autosomal dominant benign
osteopetrosis (ADO) forms (1). All
these forms are characterized by an increased bone density, which
may result in various phenotypical features, including fractures,
osteomyelitis, deformity, dental abnormalities, bone marrow
impairment and cranial nerve compression. (2). Osteopetrosis is a rare condition, and
the estimated incidence of ARO is 1 in 250,000 births, while that
of ADO is 1 in 20,000 births (3,4).
Mutations in at least 10 genes have been identified
as causative in various types of osteopetrosis cases in humans
(2). However, the majority of the
osteopetrosis cases reported thus far are caused by defects in gene
products involved in the regulation of the intra- and extracellular
pH of osteoclasts. Two significant molecules that are involved in
the acidification machinery are the proton pump, vacuolar ATPase
(V-ATPase), and the chloride-specific ion channel, CLCN7. Defects
in TCIRG1, which encodes the V-ATPase a3 subunit, have been
reported to be responsible for ARO in >50% of patients (5–7).
However, mutations in the CLCN7 gene give rise to the
complete spectrum of osteopetrosis, underlying ~15% of all ARO
cases, almost all known cases of IAO and 75% of ADO type II cases
(ADO II; Albers-Schönberg disease; MIM 166600) (8–10).
Materials and methods
Patient
A 21-month-old male was admitted to Shanghai
Children’s Medical Center (Shanghai, China) due to persistent
anemia and a lack of dentition. The patient was full-term at birth,
with a normal weight and length, and a subsequent weight and length
of 12 kg (50th percentile) and 84.8 cm (between 25th and 50th
percentile), respectively, at 21 months. A prominent forehead,
enlarged abdomen with moderate hepatosplenomegaly and visual
disturbance were noted during the physical examination. Laboratory
tests revealed that the patient had moderate anemia, elevated
levels of serum alkaline phosphatase, parathyroid hormone, creatine
kinase and MB isoenzyme, and decreased serum Ca2+
levels. The levels of phosphonium, 1,25-dihydroxy vitamin D3,
lactate dehydrogenase, thyroid hormone and thyroid stimulating
hormone were within the normal ranges. Imaging examinations
consisting of computerized tomography (CT) scans (LightSpeed 16
Slice CT; GE Healthcare, Fairfield, CT, USA) and X-rays (AMX IV
Plus Portable X-Ray, GE Healthcare) revealed a general increase of
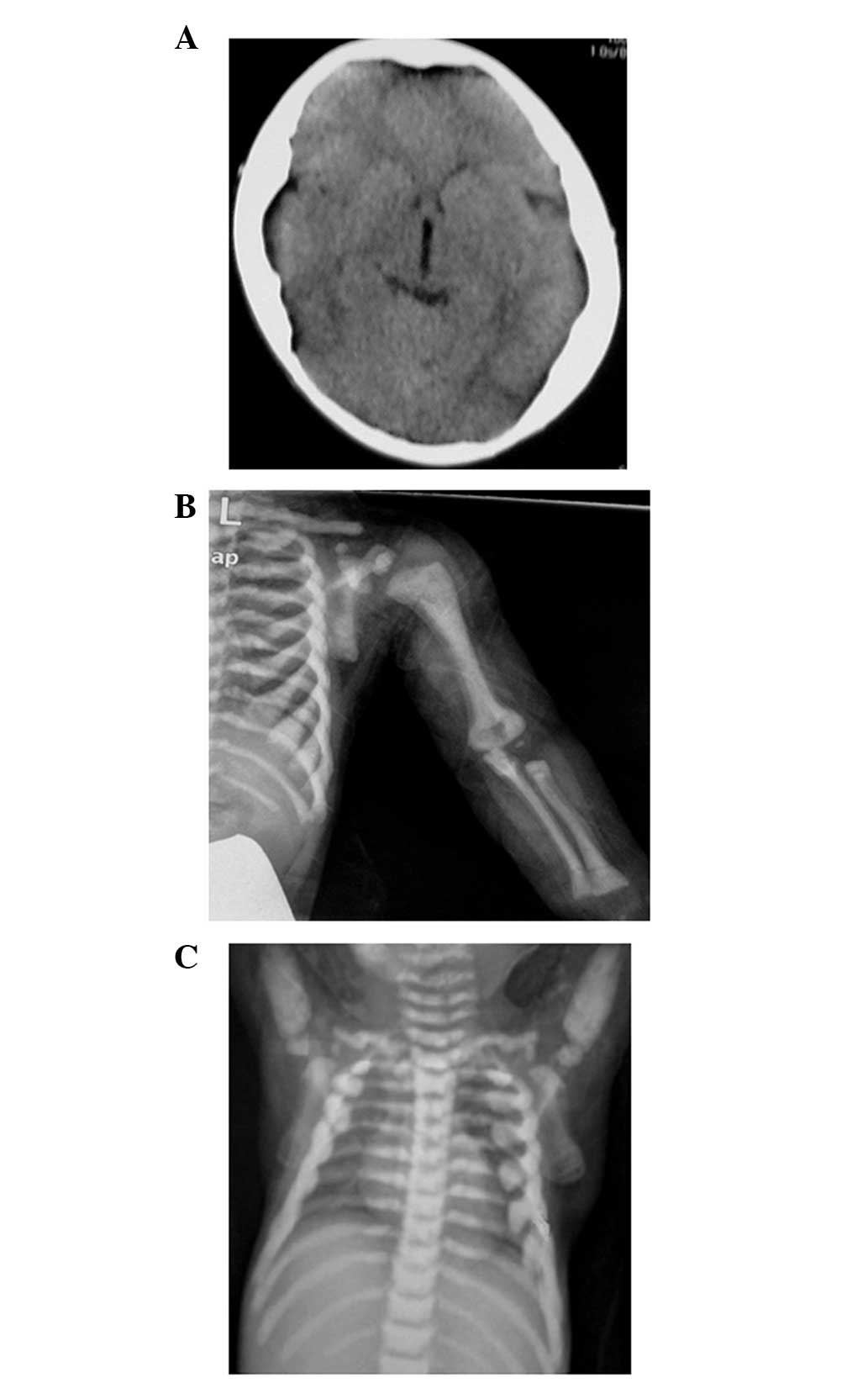
bone density involving the skull, vertebrae and limbs (Fig. 1). The diagnosis of osteopetrosis
was based on the skeletal radiographs along with the clinical and
laboratory data. The patient was scheduled to undergo a bone marrow
transplant; however, died of an infection following intensive
chemotherapy. The patient was the only son of a non-related couple.
No clinical abnormalities were noted in the parents, and further
biochemical and radiological examinations of the patient’s mother
also appeared normal.
Molecular analysis
Ethylenediaminetetraacetic acid-peripheral blood
samples were obtained from the patient, his parents and 100 healthy
individuals. Genomic DNA was isolated from the peripheral blood
leukocytes using the QIAmp DNA Blood kit (Qiagen, Hilden, Germany).
All the exons and exon-intron boundaries of the TCIRG1 and
CLCN7 genes from the patient’s genomic DNA were amplified by
PCR, and the primer sequences are listed in Tables I and II. The PCR products were analyzed by
direct DNA sequencing on an ABI 3700 sequencer (Applied Biosystems,
Foster City, CA, USA). Only genomic fragments containing the
mutations identified in the patient were amplified and sequenced
for the parents and the normal controls. Several polymorphisms,
including single-nucleotide polymorphisms (SNPs), rs12926089,
rs12926669 and rs960467, and the variable number tandem repeat
(VNTR) in intron 8 of CLCN7 were also analyzed in the
pedigree.
 | Table IPCR primers used for amplification of
TCIRG1 gene. |
Table I
PCR primers used for amplification of
TCIRG1 gene.
| Primer | Sequence (5′ to
3′) | Size (bp) |
|---|
| EXON1 F |
TCAACCTCTCCCAGACTTCC | 320 |
| EXON1 R |
CTGAGCTGCATTCACGGAG | |
| EXON2 F |
TCAGTGAGTGAAGGTGCACAG | 319 |
| EXON2 R |
GTTCAAATGGGGCCAGG | |
| EXON3 F |
TCCACACCTTTCTGGAGGAG | 266 |
| EXON3 R |
TTTCAGATCAAACTTGGCCC | |
| EXON4+EXON5 F |
GAGTTTGGGGCAGCAGG | 564 |
| EXON4+EXON5 R |
CACTGGACAAGGAGTCGGAG | |
| EXON6+EXON7 F |
GAGGCCTCCTGCCTTCC | 561 |
| EXON6+EXON7 R |
GGCCAGAAGGACACAGCTAC | |
| EXON8 F |
CCTATCGTGACTCCTCCCC | 262 |
| EXON8 R |
ACCTCCTGCACCCACCTC | |
| EXON9 F |
GAGGTGGGTGCAGGAGG | 372 |
| EXON9 R |
CTGGAAGTGAGGCAGAAACG | |
| EXON10 F |
ATCTCCAGCTGGGCCTG | 301 |
| EXON10 R |
CCTCAGGCTCACACCCAC | |
| EXON11+EXON12 F |
AAGTGATGGGTTCTTGACTGC | 570 |
| EXON11+EXON12 R |
AGGAATGCATCACTGCGG | |
| EXON13 F |
AGTCTGGCTGGAGGTGAGG | 291 |
| EXON13 R |
CACACAGGAGTGCTCAGCG | |
| EXON14+EXON15
F |
GGGAAAACAGGGTGGTGAG | 597 |
| EXON14+EXON15
R |
GATCTTGCAGCTCCCAGTG | |
| EXON16+EXON17
F |
CGTGACTGCTGTGACTCAGG | 604 |
| EXON16+EXON17
R |
GCAGAACTCGATGGTGTGG | |
| EXON18 F |
GCCTGGATGATGAAGAGGAG | 307 |
| EXON18 R |
AACTGAGGCCCAGAGAGAAG | |
| EXON19+EXON20
F |
AAGTGGGACTGTCCAAGGAG | 613 |
| EXON19+EXON20
R |
TCCCAGATCCTACACCATGC | |
| Table IIPCR primers used for amplification of
CLCN7 gene. |
Table II
PCR primers used for amplification of
CLCN7 gene.
| Primer | Sequence (5′ to
3′) | Size (bp) |
|---|
| Promoter F
(rs960467) |
GGAAGCCTCCACTCCGACCC | 475 |
| Promoter R
(rs960467) |
GTGATGAGCGACGGCGACCA | |
| Exon1 F |
CGTTGCAGGTCACATGGTC | 470 |
| Exon1 R |
GCCTCCGAAGACTCCAGAC | |
| Exon2 F |
CGGATCAGTTCTGCTTCCAG | 511 |
| Exon2 R |
CATGCTGTCACTGCTGTCCT | |
| Exon3+Exon4 F |
TGCTGGGATTGTAGGTGTCA | 629 |
| Exon3+Exon4 R |
GAGCAGCCTTCTTGGTTACG | |
| Exon5+Exon6 F |
CACACTGGGCCCTTCATAAT | 810 |
| Exon5+Exon6 R |
TCTGCTCCTCCTGAGGTTGT | |
| Exon7 F |
GTGTCTGCTGCTCTCCTCAG | 243 |
| Exon7 R |
GCTCCTGAACCAGCAAAGAG | |
| Exon8+Exon9 F (VNTR
in intron 8) |
GCTTGGCTGCTGTTTAGCTC | 764 |
| Exon8+Exon9 R (VNTR
in intron 8) |
AAGCCCATCTCCCTGAGTG | |
| Exon10+Exon11
F |
GTGCTGACCCTGCTGTCTCT | 797 |
| Exon10+Exon11
R |
AGGACCAAGGCCTGACAGA | |
| Exon12 F |
CACTGGCAAGTCCAGAGAGG | 559 |
| Exon12 R |
GCAGCAACTGTGTGACATCC | |
| Exon13 F |
CCAGTGTGTTTCTCCCCTGT | 443 |
| Exon13 R |
CTGTGGTTTTTGCCAACAGA | |
| Exon14 F |
ATTGCTCTGCTGGACACCTT | 551 |
| Exon14 R |
GCAGGGCCTCACTTCCTAC | |
| Exon15 F
(rs12926089, rs12926669) |
CAGTGTCCTCCATCAGGGACT | 401 |
| Exon15 R
(rs12926089, rs12926669) |
CTCTGAGATCTGGGTGGACAG | |
| Exon16 F |
CTCCCAACGTGTGCTCTCTC | 306 |
| Exon16 R |
ATCCTCCTGCCTTGGTCTCT | |
| Exon17 F |
TGAGAACAGGGAGCCTTCTG | 432 |
| Exon17 R |
AGGTGCGACACTTTTGTCCT | |
| Exon18+Exon19
F |
GGTGACTGTGCCCTCTGC | 730 |
| Exon18+Exon19
R |
CCCAGAAACCCTGAGCCTAC | |
| Exon20+Exon21
F |
CTGTGAGCCTCCAAACAGC | 717 |
| Exon20+Exon21
R |
GTCCACACAGCCCTCCAT | |
| Exon22+Exon23
F |
AGGCTGGTGTGAGCAGGTAG | 638 |
| Exon22+Exon23
R |
GCCCCTTGACTTCAGCTCTA | |
| Exon24+Exon25
F |
CTGAAGTCAAGGGGCTGAGG | 806 |
| Exon24+Exon25
R |
AGACCACTGCCCACAACAG | |
Reverse transcription-polymerase chain
reaction (RT-PCR)
The total RNA of the patient and the parents was
isolated from their peripheral leukocytes using the QIAmp RNA Blood
kit (Qiagen). The first-strand cDNA was synthesized using random
primers, oligo primers and PrimeScript Reverse Transcriptase
(Takara Biotechnology (Dalian) Co., Ltd., Dalian, China). The
synthesized products were amplified with primer P1,
5′-TACGGGCTCACGGTGTCTG-3′, which is located in exon 17, and primer
P2, 5′-GGTAGGCGTCTCGGAAGTC-3′, which is located in exon 23 of
CLCN7. The PCR products were further amplified using
semi-nested oligonucleotide primers P1 and P3,
5′-TTGTGCTTTAGGAGAACGA-3′, which is located in exon 22 of
CLCN7. The products were cloned into the pMD 18-T vector
(Takara Biotechnology (Dalian) Co., Ltd.) and 30 clones were
selected and sequenced.
Ethics statement
The study was approved by the Ethics Committee of
Shanghai Children’s Medical Center, Shanghai Jiao Tong University
School of Medicine. Written informed consent was obtained from the
parents and the 100 healthy individuals prior to blood sampling and
DNA analysis.
Results
Molecular analysis of the TCIRG1 and
CLCN7 genes
Direct genomic DNA sequencing of the patient
revealed compound heterozygous mutations, c.909C>A (p.Tyr303X)
and c.2008C>T (p.Arg670X), in TCIRG1 and a heterozygous
splice site mutation, c.1798-1G>T, in the CLCN7 gene
(Fig. 2). The patient’s mother
carried the TCIRG1 c.909C>A (p.Tyr303X) and CLCN7
c.1798-1G>T mutations, while the patient’s father carried the
TCIRG1 c.2008C>T (p.Arg670X) mutation. The CLCN7
c.1798-1G>T variation was not detected in the 100 healthy
individuals. In addition, several SNPs, including rs12926089,
rs12926669 and rs960467, and the intron 8 VNTR in the CLCN7
gene were investigated. However, no differences in the investigated
polymorphisms were identified between the patient and his mother
(Table III).
| Table IIIMolecular analysis of the
TCIRG1 and CLCN7 genes. |
Table III
Molecular analysis of the
TCIRG1 and CLCN7 genes.
| Subject | Nucleotide change
of TCIRG1a | Nucleotide change
of CLCN7b | Genotypesc |
|---|
|
|---|
| rs960467 | rs12926089 | rs12926669 | Intron 8 VNTR |
|---|
| Patient | c.909C>A | c.1798-1G>T | G/A | G | T | Three repeat
units |
| c.2008C>T | | | | | |
| Mother | c.909C>A | c.1798-1G>T | G/A | G | T | Three repeat
units |
| Father | c.2008C>T | -d | G | G | T | Three repeat
units |
Transcription experiment of the CLCN7
c.1798-1G>T mutation
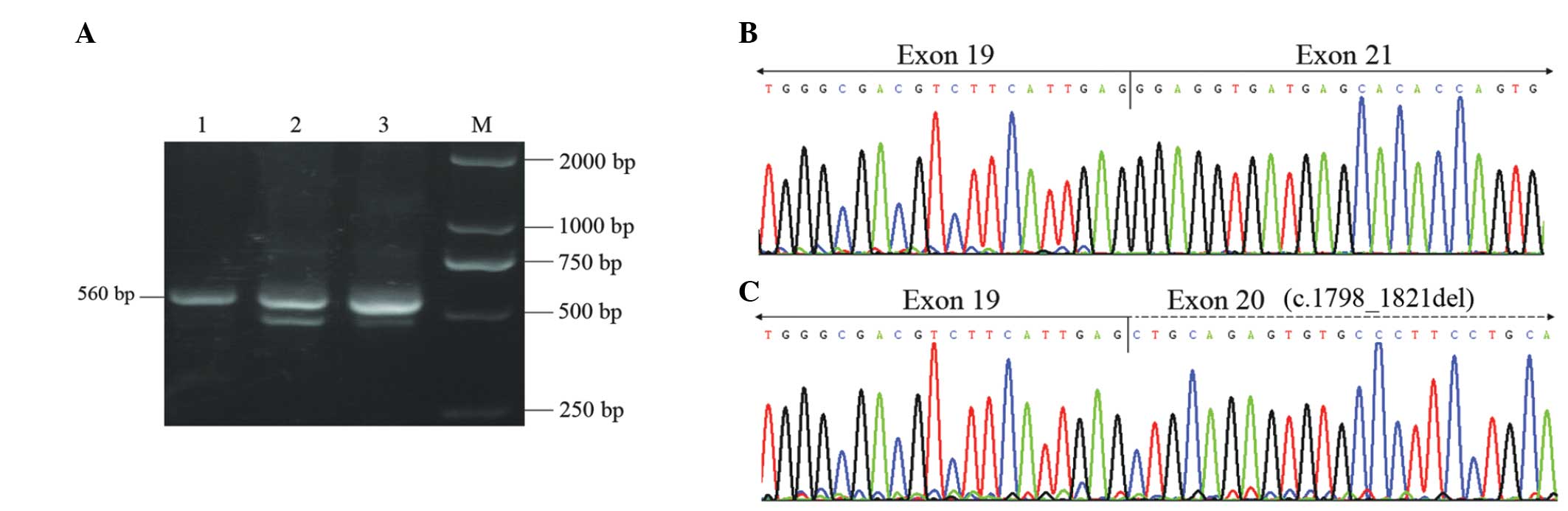
RT-PCR performed on the RNA extracted from the
father enabled the detection of one band of the expected 560 bp
(from exon 17 to exon 22 of the CLCN7 gene; Fig. 3A, lane 1). However, the same
amplification conducted using RNA extracted from the mother and the
patient, resulted in two different PCR fragments (Fig. 3A; lanes 2 and 3). In the patient,
cloning and sequencing revealed the presence of the normal
transcript corresponding to the 560-bp fragment and of two aberrant
forms of the transcripts. The majority of transcripts resulted from
the entire deletion of exon 20 (c.1798_1883del; Fig. 3B), which was detected in 11 out of
the 30 clones selected. The minority of transcripts resulted from
the deletion of the first 24 bp of exon 20 (c.1798_1821del;
Fig. 3C), which was detected in
three out of the 30 clones picked. Similar deletions were also
identified in the patient’s mother, while no aberrant transcripts
were detected in the father.
Discussion
In the present study, the case of a patient with
osteopetrosis was investigated. Molecular analysis of the patient
revealed compound heterozygous nonsense mutations in TCIRG1
and a heterozygous splice site mutation in CLCN7. Among the
three mutations, TCIRG1 c.909C>A (p.Tyr303X) and
CLCN7 c.1798-1G>T were novel mutations. CLCN7
encodes the chloride-specific ion channel, CLCN7, which cooperates
with the gene product of TCIRG1, the a3 subunit of V-ATPase
(11). CLCN7 is essential for
efficient proton pumping due to its role in neutralizing current,
and is involved in the secretion of acid into the resorption
lacuna, a specialized acidic compartment for mineral bone matrix
degradation (12,13). To the best of our knowledge, this
is the first reported case of osteopetrosis that carried the
TCIRG1 and CLCN7 gene mutations.
In order to determine the effect of CLCN7
c.1798-1G>T, a transcription experiment was subsequently
conducted. The mutations that affect mRNA splicing have been
revealed to account for a number of hereditary disorders (14–16).
Three splicing mutations of CLCN7 have been reported in
osteopetrosis thus far, including c.916+57A>T,
c.1617+6_1617+7delTG and c.2250+1G>A (9,17).
The splicing mutation, CLCN7 c.1798-1G>A, reported in the
present study has eradicated the invariant G of the AG splice
acceptor site of intron 19. The most common consequence of splicing
mutations is exon skipping, followed by the activation of aberrant
splice sites and intron retention (18,19).
In the present study, two aberrant patterns of transcripts were
detected with different proportions. The most common transcript was
the skipping of exon 20 (c.1798_1883), which was predicted to cause
a frameshift and a premature termination codon (p.Leu601GlyfsX13).
The least common was the activation of aberrant splice sites
(c.1798_1821 deletion), which was predicted to cause the in-frame
deletion of eight amino acid residues (p.Gly600_Gln607del).
Notably, aberrant splicing is predicted to affect only the
C-terminal cytosolic portion of the ClCN7 protein and not its
transmembrane portion.
Mutations in the CLCN7 gene have been
demonstrated to be involved in the pathogenesis of various forms of
osteopetrosis since 2001 (12).
Heterozygous mutations in CLCN7 may lead to ADO II, which is
associated with less severe clinical features and late onset. To
further investigate whether the CLCN7 c.1798-1G>T
mutation was pathogenic, a thorough biochemical and radiological
examination was performed on the 29-year-old mother who harbored
the heterozygous CLCN7 splicing and TCIRG1 p.Tyr303X
mutations. However, no abnormal clinical, biochemical or
radiological manifestations were observed. Previous studies have
reported several polymorphisms in the CLCN7 gene, including
rs960467, rs12926089 (Val418Met) and rs12926669, and a VNTR in
intron 8, which were associated with the penetrance of the ADO
phenotype and the variation in bone mineral density (20–22).
To determine whether these polymorphisms were associated with the
different severities of the osteopetrosis in the pedigree, the SNPs
and the VNTR were genotyped. No difference was revealed between the
patient and the mother. The CLCN7 c.1798-1G>T mutation
appeared to be a non-pathogenic variation in the present case,
although it was associated with aberrant splicing. However,
completely excluding the pathogenicity of CLCN7
c.1798-1G>T is inappropriate due to the incomplete penetrance of
ADO II. The factors involved in the phenotypic variability remain
unknown.
According to previous studies, the compound
heterozygous nonsense mutations of TCIRG1 were enough to
cause malignant osteopetrosis (5–7).
Biallelic mutations in the TCIRG1 gene are well known to be
responsible for ARO, and the absence of symptoms in the mother
together with the characteristic ARO phenotype (early postnatal
onset, generalized increased bone density and severe clinical
course, including anemia, hypodontia and visual impairment) of the
patient may indicate a diagnosis of classic TCIRG1-dependent
ARO in this case.
In conclusion, a patient with ARO was studied. The
compound heterozygous mutations, c.909C>A (p.Tyr303X) and
c.2008C>T (p.Arg670X), in TCIRG1 and a heterozygous
splicing mutation, c.1798-1G>T, in the CLCN7 gene were
identified in the patient. Among the three mutations, TCIRG1
c.909C>A (p.Tyr303X) and CLCN7 c.1798-1G>T were novel
mutations. This study highlights that TCIRG1 and
CLCN7 should be sequenced in order to gain a comprehensive
molecular diagnosis of osteopetrosis.
Acknowledgements
The authors would like to thank all the members of
the family for their participation in this study. This study was
supported by the National Natural Science Foundation of China
(grant nos. 81000207 and 81000346) and a foundation grant from
Shanghai Science and Technology Commission for major issues (grant
no. 11dz1950300).
References
|
1
|
Tolar J, Teitelbaum SL and Orchard PJ:
Osteopetrosis. N Engl J Med. 351:2839–2849. 2004. View Article : Google Scholar
|
|
2
|
Stark Z and Savarirayan R: Osteopetrosis.
Orphanet J Rare Dis. 4:52009. View Article : Google Scholar
|
|
3
|
Loría-Cortés R, Quesada-Calvo E and
Cordero-Chaverri C: Osteopetrosis in children: a report of 26
cases. J Pediatr. 91:43–47. 1977.
|
|
4
|
Bollerslev J and Andersen PE Jr:
Radiological, biochemical and hereditary evidence of two types of
autosomal dominant osteopetrosis. Bone. 9:7–13. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Frattini A, Orchard PJ, Sobacchi C,
Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK,
Wallbrandt P, Zecca L, et al: Defects in TCIRG1 subunit of the
vacuolar proton pump are responsible for a subset of human
autosomal recessive osteopetrosis. Nat Genet. 25:343–346. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kornak U, Schulz A, Friedrich W, Uhlhaas
S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ and Kubisch C:
Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause
infantile malignant osteopetrosis. Hum Mol Genet. 9:2059–2063.
2000.
|
|
7
|
Sobacchi C, Frattini A, Orchard P, Porras
O, Tezcan I, Andolina M, Babul-Hirji R, Baric I, Canham N, Chitayat
D, et al: The mutational spectrum of human malignant autosomal
recessive osteopetrosis. Hum Mol Genet. 10:1767–1773. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cleiren E, Bénichou O, Van Hul E, Gram J,
Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama
T, et al: Albers-Schönberg disease (autosomal dominant
osteopetrosis, type II) results from mutations in the ClCN7
chloride channel gene. Hum Mol Genet. 10:2861–2867. 2001.
|
|
9
|
Frattini A, Pangrazio A, Susani L,
Sobacchi C, Mirolo M, Abinun M, Andolina M, Flanagan A, Horwitz EM,
Mihci E, et al: Chloride channel ClCN7 mutations are responsible
for severe recessive, dominant, and intermediate osteopetrosis. J
Bone Miner Res. 18:1740–1747. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Del Fattore A, Peruzzi B, Rucci N, Recchia
I, Cappariello A, Longo M, Fortunati D, Ballanti P, Iacobini M,
Luciani M, et al: Clinical, genetic, and cellular analysis of 49
osteopetrotic patients: implications for diagnosis and treatment. J
Med Genet. 43:315–325. 2006.
|
|
11
|
Supanchart C and Kornak U: Ion channels
and transporters in osteoclasts. Arch Biochem Biophys. 473:161–165.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kornak U, Kasper D, Bösl MR, Kaiser E,
Schweizer M, Schulz A, Friedrich W, Delling G and Jentsch TJ: Loss
of the ClC-7 chloride channel leads to osteopetrosis in mice and
man. Cell. 104:205–215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Väänänen HK, Zhao H, Mulari M and Halleen
JM: The cell biology of osteoclast function. J Cell Sci.
113:377–381. 2000.
|
|
14
|
Ars E, Serra E, García J, Kruyer H, Gaona
A, Lázaro C and Estivill X: Mutations affecting mRNA splicing are
the most common molecular defects in patients with
neurofibromatosis type 1. Hum Mol Genet. 9:237–247. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
López-Bigas N, Audit B, Ouzounis C, Parra
G and Guigó R: Are splicing mutations the most frequent cause of
hereditary disease? FEBS Lett. 579:1900–1903. 2005.PubMed/NCBI
|
|
16
|
Yu T, Wang X, Ding Q, Fu Q, Dai J, Lu Y,
Xi X and Wang H: Using a minigene approach to characterize a novel
splice site mutation in human F7 gene causing inherited factor VII
deficiency in a Chinese pedigree. Haemophilia. 15:1262–1266. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pangrazio A, Pusch M, Caldana E, Frattini
A, Lanino E, Tamhankar PM, Phadke S, Lopez AG, Orchard P, Mihci E,
et al: Molecular and clinical heterogeneity in CLCN7 dependent
osteopetrosis report of 20 novel mutations. Hum Mutat.
31:E1071–E1080. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krawczak M, Reiss J and Cooper DN: The
mutational spectrum of single base-pair substitutions in mRNA
splice junctions of human genes: causes and consequences. Hum
Genet. 90:41–54. 1992. View Article : Google Scholar
|
|
19
|
Nakai K and Sakamoto H: Construction of a
novel database containing aberrant splicing mutations of mammalian
genes. Gene. 141:171–177. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chu K, Koller DL, Snyder R, Fishburn T,
Lai D, Waguespack SG, Foroud T and Econs MJ: Analysis of variation
in expression of autosomal dominant osteopetrosis type 2: searching
for modifier genes. Bone. 37:655–661. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pettersson U, Albagha OM, Mirolo M,
Taranta A, Frattini A, McGuigan FE, Vezzoni P, Teti A, van Hul W,
Reid DM, Villa A and Ralston SH: Polymorphisms of the CLCN7 gene
are associated with BMD in women. J Bone Miner Res. 20:1960–1967.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kornak U, Ostertag A, Branger S, Benichou
O and de Vernejoul MC: Polymorphisms in the CLCN7 gene modulate
bone density in postmenopausal women and in patients with autosomal
dominant osteopetrosis type II. J Clin Endocrinol Metab.
91:995–1000. 2006. View Article : Google Scholar : PubMed/NCBI
|