Introduction
Alzheimer’s disease (AD) is the most prevalent
neurodegenerative disease affecting >25 million people
worldwide. It is characterized by progressive loss of cognitive
function resulting in dementia and mortality (1). The pathological hallmarks of AD are
extracellular senile plaques (SPs) composed of amyloid β (Aβ) and
intracellular neurofibrillary tangles consisting of
hyperphosphorylated microtubule-associated protein τ (2). Aβ aggregation and accumulation,
derived from sequential cleavage of amyloid precursor protein (APP)
mediated by β- and γ-secretases, is the triggering event in the
process of AD (2–4). The Aβ oligomer is the main form which
mediates the deleterious effect of Aβ as it fibrillates to form
SPs. Emerging evidence has indicated the possible interaction
between Aβ and τ, and their synergistic effects during AD
progression (5). However, the
underlying molecular mechanisms linking Aβ and τ remain poorly
understood.
Synaptic dysfunction has also been detected in the
brains of patients with AD prior to the appearance of amyloid
plaques (6). Aβ42 oligomers have
been reported to induce the acute rapid synaptotoxic effect and τ
phosphorylation at Ser-262 by activating the
calcium/calmodulin-dependent protein kinase kinase 2, adenosine
monophosphate-activated protein kinase (CAMKK2-AMPK) pathway
(1). AMPK acts as a metabolic
sensor and is an essential regulator of the cellular metabolism.
AMPK is activated by an increased intracellular AMP/adenosine
triphosphate (ATP) ratio as well as other forms of cellular stress.
Once activated, AMPK regulates a variety of biological processes,
including cell polarity, apoptosis, cell migration and synaptic
plasticity (7,8). Accumulating studies have indicated
that AMPK-signaling regulates τ phosphorylation and amyloidogenesis
in the AD pathogenesis (1,9–11).
Activated AMPK was observed to markedly enriched in tangle-bearing
neurons in patients with AD (12).
These observations indicate that AMPK may be involved in the
pathogenesis of AD.
microRNAs (miRNAs) are short, non-coding RNAs that
inhibit protein expression by binding to specific recognition
elements in the 3′ untranslated region (3′UTR) of target
transcripts leading to mRNA translation suppression or mRNA
degradation. Currently, >700 miRNAs have been identified, and
are essential in a number of cellular processes, including cell
polarity, migration, apoptosis and synaptic plasticity (13). miRNA expression profiles in
patients with AD and AD animal models have been identified. miRNAs
have been reported to regulate Aβ generation, the inflammatory
response and neurogenesis in AD pathogenesis (14).
miR-9 is a synapse-enriched miRNA and was observed
to be markedly decreased in patients with AD (15). miR-9 is specifically expressed in
the brain and promotes neurogenesis by suppressing the basic
helix-loop-helix hairy/enhancer of split-1 [E(sp1)] transcription
factors Her5 and Her9 expression (16). Schonrock et al reported that
miR-9 was downregulated by Aβ treatment and suppressed the
expression of a variety of genes (17,18).
However, the role of miR-9 during the Aβ-induced synaptotoxic
effect is poorly understood. In the present study the effect of
miR-9 on Aβ42-triggered CAMKK2-AMPK activation and the synaptotoxic
impairment was investigated.
Materials and methods
Aβ42 oligomer preparation
Aβ42 oligomer preparation was performed as
previously reported (1). Briefly,
the Aβ42 peptides (China Peptide, Shanghai, China) were dissolved
in hexafluoro-2-propanol (HFIP) for 2 h, and then the HFIP was
removed by speed vacuum (Neu-Tec Group Inc., Farmingdale, NY, USA).
Dimethylsulfoxide was added to produce a 5 mM solution. This
solution was added to cold phenol red-free F12 medium (Invitrogen
Life Technologies, New York, NY, USA), incubated at 4°C for 24 h
and then centrifuged at 14,000 × g for 10 min to discard fibrils.
The supernatant was kept and used as a source of Aβ42
oligomers.
RNA extraction and quantitative PCR
(qPCR)
The total RNA was collected from cells with TRIzol
(Invitrogen Life Technologies), according to the manufacturer’s
instructions. In total, 1 μg RNA was reverse-transcribed into cDNA
with a reverse transcription kit (Toyobo, Dalian, China). miRNAs
were collected using a microRNA Extraction kit (Tiangen, Beijing,
China). Poly(A) was added and 1 μg RNA containing miRNAs was
reversely transcribed into cDNA. The synthesized cDNAs were
amplified using the SYBR qPCR kit (Takara, Dalian, China) on ABI
Stepone plus equipment (ABI, Foster City, CA, USA). Expression of
CAMKK2 was normalized with GAPDH, and miR-9 and miR-181c levels
were normalized with U6 snRNA.
Constructs and luciferase assay
The miR-9 expression construct (catalog no.
MmiR-AN0825-AM02) was purchased from GeneCopoeia (Rockville, MD,
USA). The possible target positions of the CAMKK2 3′UTR sequences
were subcloned into the psiCHECK-2 between XhoI and
NotI restriction sites (Promega Corporation, Madison, WI,
USA). The position 1898-2358 of CAMKK2 3′UTR was amplified from
mouse hippocampus cDNA with the following primers: Sense:
5′-CTCGAGTGCCCGAGTAGGGTAGGCGTG-3′ and antisense:
3′-AGCGGCCGCTGAACGAGGCTTGTGCTT-5′. Mutations in the miR-9
binding-sites of CAMKK2 were introduced with a fast whole-plasmid
mutation kit (NEB, Ipswich, Canada).
HEK-293 cells were plated onto a 96-well plate.
Subsequent to 24 h incubation, the cells were treated with a
cotransfection consisting of 35 μl serum-free medium, 0.5 μl
Lipofectamine 2000, 0.03 μg psiCHECK-2-CAMKK2 and 0.1 μg miR-9 per
well. Renilla luciferase or pEZX-AM02 vector was used as a
negative control. After 4 h, 100 μl serum-containing culture medium
was added to the wells. The luciferase activity was examined 48 h
after transfection using the Dual-Luciferase®Reporter
1000 Assay system (Promega Corporation).
Primary neuronal culture and
transfection
Primary embryonic E18 hippocampal neurons of the
mice were cultured according to a procedure described previously
(19). Briefly, the hippocampal
neurons were collected and incubated with 5 ml D-Hank’s containing
0.25% trypsin for 15 min and centrifuged at 1000 × g for 5 min
following addition of 5 ml Dulbecco’s modified Eagle’s medium with
Ham’s F12 medium with 10% fetal bovine serum, the cells were
triturated and seeded onto a 60-mm plastic culture dish at a
density of 100–200 neurons/mm2 and maintained in a
humidified incubator at 37°C with 5% CO2 for 4 h. Next,
the culture medium was replaced by neurobasal medium with 2% B27
and the cells were cultured for 18 days. Transfection was performed
according to the manufacturer’s instructions (Invitrogen Life
Technologies), and the ratio of the constructs to Lipofectamine
2000 was 1:2.
AMPK activity assay
Cellular AMPK activity was determined using the SAMS
peptide phosphorylation assay kit (Upstate Biotechnology, Lake
Placid, NY, USA) according to the manufacturer’s instructions.
Briefly, the cells were maintained in serum-free medium for 12 h
prior to drug exposure. Aβ42 oligomers or the scramble control to
the final concentration of 1 μM were added to the cell culture, and
incubated at 37°C for 1 h. The culture medium was removed, and the
cells were harvested in Tris-HCl supplemented with the protease
inhibitors aprotinin, leupeptin, and pepstain A (Roche Diagnostics,
Pleasanton CA, USA). The cellular debris were removed following
centrifugation at 10,000 × g at 4°C for 15 min and the supernatants
were stored at −70°C prior to the AMPK activity assays (20).
Western blot analysis
The cells were harvested and extracted with protein
radio-immunoprecipitation assay buffer, supplemented with a
cocktail of protease (Roche) and phosphatase (Sigma-Aldrich, SG,
Switzerland) inhibitors. Equal quantities of proteins were
separated by 10% sodium dodecyl sulfate-polyacrylamide gel,
transferred to polyvinylidene difluoride membranes (Millipore,
Billerica, MA, USA) and detected using specific primary antibodies
overnight at 4°C, including pT172 (rabbit monoclonal; Cell
Signaling Technology, Inc., San Diego, CA, USA), AMPK (rabbit
polyclonal; Cell Signaling Technology, Inc.), CAMKK2 (rabbit
polyclonal; Abcam, Cambridge, UK), GAPDH (rabbit monoclonal; Cell
Signaling), pS262 (rabbit polyclonal; Abcam) and Tau (mouse
monoclonal; Millipore, Hayward, CA, USA). The membranes were
incubated with secondary antibodies, conjugated to horseradish
peroxidase for 1 h at 37°C and visualized using an enhanced
chemiluminescence kit (Pierce, Rockford, IL, USA). The blots were
scanned and analyzed by Kodak Digital Science 1D software (Eastman
Kodak, Rochester, NY, USA).
Image acquisition and analyses
The images were acquired in 2048×2048 resolution
using the A1R laser-scanning confocal microscope (Nikon A1R-si
Laser scanning confocal microscope; Nikon, Tokyo, Japan) with the
Nikon software NIS-Elements (Nikon, Melville, NY, USA). The
dendritic spine density was quantified on branches proximal to the
soma.
Statistical analysis
Data were analyzed by using SPSS 16.0 statistical
software (SPSS Inc., Chicago, IL, USA), and one-way analysis of
variance with Dunnett’s post-test was used for multiple
comparisons. Student’s t-test was used to determine the differences
between the two groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Overexpression of miR-9 inhibited
Aβ42-induced AMPK activation
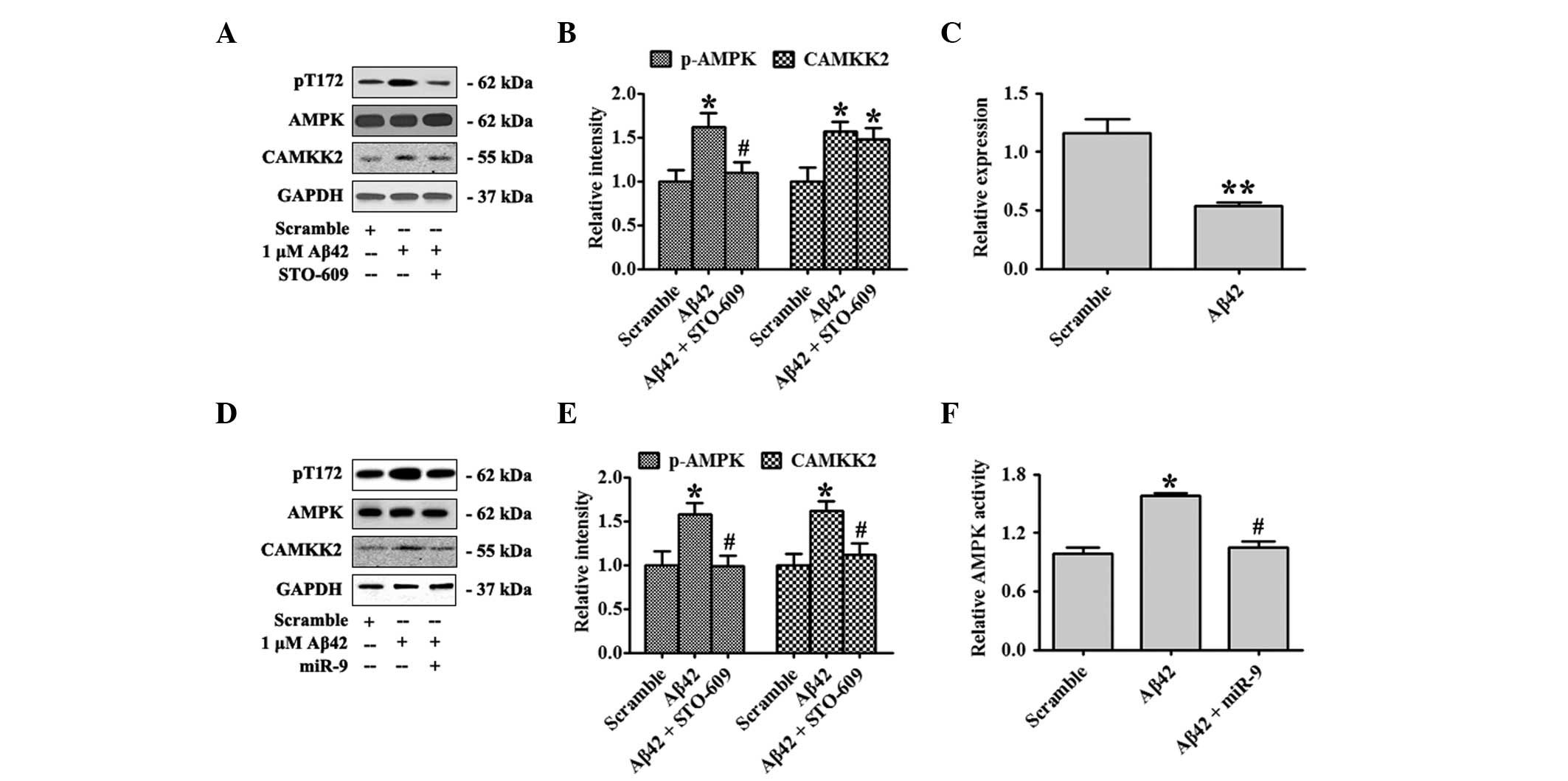
The effect of Aβ42 oligomers on the activation of
the CAMKK2-AMPK2 pathway was confirmed in the present study. Days
in vitro (DIV)18 primary hippocampal neurons were treated
with 1 μM Aβ42 oligomers or the scrambled control for 12 h.
Activated AMPK, pT172-AMPK and total AMPK were detected by western
blot analysis. Aβ42 elevated CAMKK2 expression and Aβ42 treatment
also significantly increased the ratio of pT172-AMPK/AMPK. A
supplement of STO-609, a specific inhibitor of CAMKK2, attenuated
the levels of pT172-AMPK (Fig. 1A and
B). Previous studies reported that Aβ42 could decrease the
expression of several miRNAs including miR-9. In the present study
the expression of miR-9 in primary hippocampal neurons treated with
Aβ42 oligomers for 12 h was measured. Aβ42 markedly inhibited miR-9
expression (Fig. 1C). In order to
evaluate the function of miR-9 on the CAMKK2-AMPK pathway activated
by Aβ42 oligomers, miR-9 at DIV 10 was overexpressed and neurons
were treated with Aβ42 oligomers at DIV18. miR-9 overexpression was
found to substantially eradicate the elevation of CAMKK2 expression
and the activation of AMPK as shown by decreased CAMKK2 and
pT172-AMPK levels (Fig. 1D and E).
AMPK activity was also examined by the SAMS Peptide Phosphorylation
Assay kit (Fig 1D). These data
indicated that overexpression of miR-9 inhibited CAMKK2-AMPK
activation by Aβ42 oligomers.
miR-9 suppressed CAMKK2 translation
To analyze the potential targets of miR-9, Pictar
(http://pictar.mdc-berlin.de/) and
Targetscan 6.2 (http://www.targetscan.org/) Bioinformatics’ algorithms
were used to screen the potential gene. CAMKK2 was identified to be
a potential target of miR-9 predicted by the two algorithms. The
predicted sequences are shown in Fig.
2A. To test this hypothesis, a wild-type and a mutant of CAMKK2
3′UTR were generated and these sequences were inserted into the
luciferase reporter vector. When coexpressed with miR-9, a
wild-type reporter revealed significant inhibition (Fig. 2B), while without effect on its
mutant (Mut). The mRNA and protein levels of CAMKK2 were also
detected. miR-9 significantly decreased the protein level of CAMKK2
(Fig. 2C and D), while it did not
affect the mRNA level of CAMKK2 (Fig.
2E). These results demonstrated that miR-9 directly targeted
CAMKK2.
miR-9 rescued Aβ42-induced
synaptotoxicity by targeting CAMKK2
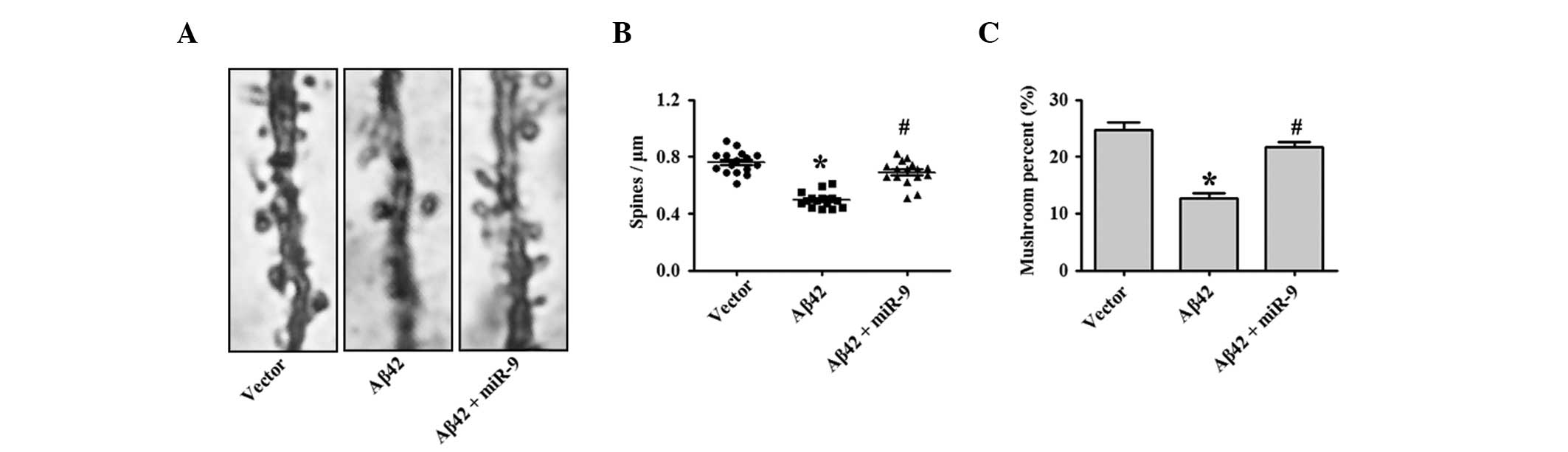
Previous studies demonstrated that 1 μM Aβ42
oligomers induced a significant reduction in dendritic spine
density, while it did not affect neuronal viability (1). In addition, the present study tested
whether miR-9 overexpression was sufficient to eradicate the
synaptotoxic effects of Aβ42 oligomers. As shown in Fig. 3A and quantified in Fig. 3B and C, the results demonstrate
that the overexpression of miR-9 restored the reduction in spine
density following Aβ42 oligomer application.
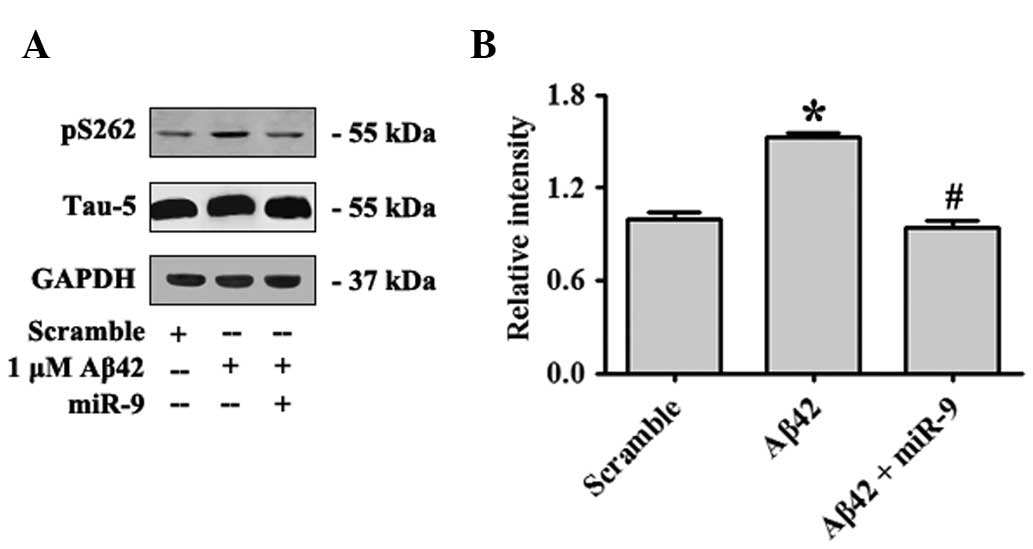
miR-9 attenuated Aβ42-induced τ
phosphorylation by targeting CAMKK2
Plaques of Aβ deposition and tangles formed by
hyperphosphorylated microtubule-binding protein τ were the two
major histopathological signatures observed in the brains of AD
patients. Although Aβ and τ have been extensively investigated
independently with regard to their separate toxic effects, recent
results indicated their potential interactions or synergistic
effects during AD progression (21,22).
For instance, τ was observed to mediate Aβ signals to drive ectopic
neuronal cell cycle re-entry in AD (23). Recent results had revealed that
AMPK is a potent τ kinase (24).
S262 of τ was a significant target of AMPK, and Aβ42 oligomers
increased pS262-τ phosphorylation by activating AMPK (25). In the present study, overexpression
of miR-9 was identified to attenuate Aβ42-induced τ phosphorylation
by targeting CAMKK2 (Fig. 4A and
B).
Discussion
Loss of synapses begins during the early stages of
AD, prior to plaque formation, and progressively affect neuronal
activity, leading to cognitive impairment. Aβ oligomers had been
hypothesized to contribute to synapse loss, and τ acted as an
essential mediator of Aβ synaptotoxicity. In vitro and in
vivo studies demonstrated that Aβ42 oligomers activated the
CAMKK2-AMPK pathway, which phosphorylated τ on S262 in the
microtubule-binding domain inducing dendritic spine loss in
hippocampal neurons (1,24,26).
Inhibition of CAMKK2, or overexpression of the unphosphorylated
mutant of τ (S262A) eradicated Aβ42 oligomer-induced
synaptotoxicity (1,27).
AMPK is a sensor of cellular stress, which maintains
energy homeostasis by regulating several metabolic enzyme
activities. AMPK is a heterotrimeric Ser/Thr kinase, with a
catalytic α subunit and two regulatory subunits, β and γ.
Regulation of AMPK activity involved activation by AMP and
phosphorylation of the AMPKα subunit at Thr-172 within the
activation loop by its upstream kinases. The major upstream kinases
included liver kinase B1, in response to increased AMP/ATP ratio,
and CAMKK2, in response to elevated intracellular Ca2+
levels (28). By contrast, Thr-172
was dephosphorylated by protein phosphatase-2C to deactivate AMPK.
AMPK was widely expressed in mammalian tissues and cell types,
including the hippocampus. Epidemiological studies and functional
neuroimaging have demonstrated perturbed brain energy metabolism in
patients with AD. Perturbation of brain energy metabolism is
involved in the neurodegeneration occurring in early stages of AD,
and may correlate with early cognitive dysfunction, including
increased problems in maintaining Ca2+ homeostasis,
decline in glucose uptake, synaptotoxicity and mitochondrial
dysfunction (29). AMPK also
regulated Aβ generation through regulating APP processing (30).
miRNAs are significant in a variety of neurological
processes, including synaptic plasticity and stress responses.
Previous studies of miRNAs in AD demonstrated that numerous miRNAs,
including miR-9, miR-124a, miR-125b and miR-132, abundantly
expressed in fetal hippocampus were differentially regulated in the
aged brain (31). miRNAs were also
shown to participate in the pathogenesis of AD. For instance,
miR-29a/b-1, miR-195, miR-298 and miR-328 suppressed Aβ generation
by inhibiting a β-amyloid precursor protein-converting enzyme
(13,32,33).
miR-16, miR-17-5p, miR-20a, miR-106a and miR-106b were reported to
regulate APP expression, indicating that variations in miRNA
expression may contribute to the alteration in APP expression and
Aβ production in the brain during development and disease (34,35).
Conversely, Aβ also induced abnormal expression of miRNAs,
including miR-9, miR-21 and miR-181c (18). However, the role of miRNAs in Aβ
induced synaptotoxic effect remains poorly understood.
miR-9, a brain-specific and synapse enriched miRNA,
was significantly decreased in patients with AD (36). miR-9 was initially identified as a
crucial regulator of the development and physiology of the nervous
system in numerous organisms, including Drosophila and
mammals (16). miR-9 regulated the
proliferation, differentiation and migration of neural stem cells
by controlling hairy/E (spl1) (37). miR-9 was able to improve neurite
outgrowth by targeting Forkhead box protein P2 (37). Although miR-9 was decreased in
response to Aβ42, the potential role of miR-9 in Aβ42 induced
synaptotoxicity remains unknown. In the present study
overexpression of miR-9 was found to be capable of attenuating
Aβ42-induced CAMKK2-AMPK pathway activation, rescuing Aβ42-induced
dendritic spine density loss and eradicating Aβ42-induced τ
phosphorylation on S262 partially by targeting CAMKK2.
In conclusion, miR-9 was shown to antagonize the
Aβ42-induced synaptotoxic effect by targeting CAMKK2, which may
provide a novel strategy for AD therapy.
References
|
1
|
Mairet-Coello G, Courchet J, Pieraut S,
Courchet V, Maximov A and Polleux F: The CAMKK2-AMPK kinase pathway
mediates the synaptotoxic effects of Aβ oligomers through tau
phosphorylation. Neuron. 78:94–108. 2013.PubMed/NCBI
|
|
2
|
Wang JZ and Liu F: Microtubule-associated
protein tau in development, degeneration and protection of neurons.
Prog Neurobiol. 85:148–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vanitallie TB: Preclinical sporadic
Alzheimer’s disease: target for personalized diagnosis and
preventive intervention. Metabolism. 62(Suppl 1): S30–S33.
2013.
|
|
4
|
Wang JF, Lu R and Wang YZ: Regulation of β
cleavage of amyloid precursor protein. Neurosci Bull. 26:417–427.
2010.
|
|
5
|
Jin M, Shepardson N, Yang T, Chen G, Walsh
D and Selkoe DJ: Soluble amyloid beta-protein dimers isolated from
Alzheimer cortex directly induce Tau hyperphosphorylation and
neuritic degeneration. Proc Natl Acad Sci USA. 108:5819–5824. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Palop JJ, Chin J, Roberson ED, et al:
Aberrant excitatory neuronal activity and compensatory remodeling
of inhibitory hippocampal circuits in mouse models of Alzheimer’s
disease. Neuron. 55:697–711. 2007.PubMed/NCBI
|
|
7
|
Yuan HX, Xiong Y and Guan KL: Nutrient
sensing, metabolism, and cell growth control. Mol Cell. 49:379–387.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakano A and Takashima S: LKB1 and
AMP-activated protein kinase: regulators of cell polarity. Genes
Cells. 17:737–747. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salminen A, Kaarniranta K, Haapasalo A,
Soininen H and Hiltunen M: AMP-activated protein kinase: a
potential player in Alzheimer’s disease. J Neurochem. 118:460–474.
2011.
|
|
10
|
Park H, Kam TI, Kim Y, et al:
Neuropathogenic role of adenylate kinase-1 in Aβ-mediated tau
phosphorylation via AMPK and GSK3β. Hum Mol Genet. 21:2725–2737.
2012.PubMed/NCBI
|
|
11
|
Kim J, Park YJ, Jang Y and Kwon YH: AMPK
activation inhibits apoptosis and tau hyperphosphorylation mediated
by palmitate in SH-SY5Y cells. Brain Res. 1418:42–51. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vingtdeux V, Davies P, Dickson DW and
Marambaud P: AMPK is abnormally activated in tangle- and
pre-tangle-bearing neurons in Alzheimer’s disease and other
tauopathies. Acta Neuropathol. 121:337–349. 2011.PubMed/NCBI
|
|
13
|
Fernandez-Hernando C, Ramírez CM, Goedeke
L and Suárez Y: MicroRNAs in metabolic disease. Arterioscler Thromb
Vasc Biol. 33:178–185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delay C, Mandemakers W and Hebert SS:
MicroRNAs in Alzheimer’s disease. Neurobiol Dis. 46:285–290.
2012.
|
|
15
|
Hébert SS, Horré K, Nicolaï L, et al: Loss
of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease
correlates with increased BACE1/beta-secretase expression. Proc
Natl Acad Sci USA. 105:6415–6420. 2008.PubMed/NCBI
|
|
16
|
Leucht C, Stigloher C, Wizenmann A, Klafke
R, Folchert A and Bally-Cuif L: MicroRNA-9 directs late organizer
activity of the midbrain-hindbrain boundary. Nat Neurosci.
11:641–648. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schonrock N, Humphreys DT, Preiss T and
Götz J: Target gene repression mediated by miRNAs miR-181c and
miR-9 both of which are down-regulated by amyloid-β. J Mol
Neurosci. 46:324–335. 2012.PubMed/NCBI
|
|
18
|
Schonrock N, Ke YD, Humphreys D, et al:
Neuronal microRNA deregulation in response to Alzheimer’s disease
amyloid-beta. PLoS One. 5:e110702010.PubMed/NCBI
|
|
19
|
Zhu LQ, Zheng HY, Peng CX, et al: Protein
phosphatase 2A facilitates axonogenesis by dephosphorylating CRMP2.
J Neurosci. 30:3839–3848. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rencurel F, Foretz M, Kaufmann MR, et al:
Stimulation of AMP-activated protein kinase is essential for the
induction of drug metabolizing enzymes by phenobarbital in human
and mouse liver. Mol Pharmacol. 70:1925–1934. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Handoko M, Grant M, Kuskowski M, et al:
Correlation of specific amyloid-β oligomers with tau in
cerebrospinal fluid from cognitively normal older adults. JAMA
Neurol. 5:594–599. 2013.
|
|
22
|
Gao L, Tian S, Gao H and Xu Y: Hypoxia
increases Aβ-induced tau phosphorylation by calpain and promotes
behavioral consequences in AD transgenic mice. J Mol Neurosci.
51:128–147. 2013.
|
|
23
|
Seward ME, Swanson E, Norambuena A, et al:
Amyloid-β signals through tau to drive ectopic neuronal cell cycle
re-entry in Alzheimer’s disease. J Cell Sci. 126:1278–1286.
2013.
|
|
24
|
Thornton C, Bright NJ, Sastre M, Muckett
PJ and Carling D: AMP-activated protein kinase (AMPK) is a tau
kinase, activated in response to amyloid β-peptide exposure.
Biochem J. 434:503–512. 2011.
|
|
25
|
Yoshida H and Goedert M: Phosphorylation
of microtubule-associated protein tau by AMPK-related kinases. J
Neurochem. 120:165–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moolman DL, Vitolo OV, Vonsattel JP and
Shelanski ML: Dendrite and dendritic spine alterations in Alzheimer
models. J Neurocytol. 33:377–387. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Manczak M and Reddy PH: Abnormal
interaction of oligomeric amyloid-β with phosphorylated tau:
implications to synaptic dysfunction and neuronal damage. J
Alzheimers Dis. 36:285–295. 2013.
|
|
28
|
Viollet B, Lantier L, Devin-Leclerc J, et
al: Targeting the AMPK pathway for the treatment of Type 2
diabetes. Front Biosci (Landmark Ed). 14:3380–3400. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cai Z, Yan LJ, Li K, Quazi SH and Zhao B:
Roles of AMP-activated protein kinase in Alzheimer’s disease.
Neuromolecular Med. 14:1–14. 2012.
|
|
30
|
Lu J, Wu DM, Zheng YL, et al: Quercetin
activates AMP-activated protein kinase by reducing PP2C expression
protecting old mouse brain against high cholesterol-induced
neurotoxicity. J Pathol. 222:199–212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lukiw WJ: Micro-RNA speciation in fetal,
adult and Alzheimer’s disease hippocampus. Neuroreport. 18:297–300.
2007.PubMed/NCBI
|
|
32
|
Ai J, Sun LH, Che H, et al: MicroRNA-195
protects against dementia induced by chronic brain hypoperfusion
via its anti-amyloidogenic effect in rats. J Neurosci.
33:3989–4001. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boissonneault V, Plante I, Rivest S and
Provost P: MicroRNA-298 and microRNA-328 regulate expression of
mouse beta-amyloid precursor protein-converting enzyme 1. J Biol
Chem. 284:1971–1981. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu W, Liu C, Zhu J, et al: MicroRNA-16
targets amyloid precursor protein to potentially modulate
Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol Aging.
33:522–534. 2012.PubMed/NCBI
|
|
35
|
Hébert SS, Horré K, Nicolaï L, et al:
MicroRNA regulation of Alzheimer’s Amyloid precursor protein
expression. Neurobiol Dis. 33:422–428. 2009.
|
|
36
|
Cogswell JP, Ward J, Taylor IA, et al:
Identification of miRNA changes in Alzheimer’s disease brain and
CSF yields putative biomarkers and insights into disease pathways.
J Alzheimers Dis. 14:27–41. 2008.
|
|
37
|
Tan SL, Ohtsuka T, González A and Kageyama
R: MicroRNA9 regulates neural stem cell differentiation by
controlling Hes1 expression dynamics in the developing brain. Genes
Cells. 17:952–961. 2012. View Article : Google Scholar : PubMed/NCBI
|