Introduction
Our genomic DNA is constantly attacked and damaged
by normal intracellular metabolic processes and a number of
environmental factors, including ultraviolet (UV) light, ionizing
radiation (IR), radiomimetic drugs and reactive oxygen species
(ROS) (1,2). DNA damage also occurs spontaneously
during DNA replication (2). To
cope with these lesions, eukaryotic cells have evolved the complex
DNA damage response (DDR) machinery (2). DDR senses damaged DNA and activates
the cell cycle checkpoint to halt cell cycle progression, allowing
time for DNA repair. The DDR process also induces apoptosis or
senescence when damaged DNA fails to be repaired (2). Deficiency in DDR leads to mutations,
genomic instability and cellular senescence, an enabling
characteristic associated with cancer (3).
Of the various forms of DNA damage, DNA
double-strand breaks (DSBs) are the most lethal (1). There are two main pathways that are
independently responsible for DSB repair in mammalian cells, namely
non-homologous end-joining (NHEJ) and homologous recombination (HR)
(2). NHEJ is the predominant
pathway, which induces the direct ligation of two broken DNA ends
together (1,4). NHEJ repair occurs during G0 and G1
phase of the cell cycle, when the extensive sequence homology for
repair is absent (1).
The NHEJ pathway is initiated by the rapid
association of the Ku70/Ku80 heterodimer to the broken DNA ends
(2). Ku70/Ku80 then recruits the
catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs) to
form a DNA-PK complex (5). This
kinase complex phosphorylates the nuclease Artemis to facilitate
the initial processing of ends, and provides protection of the ends
required for the following DNA ligation by another complex
containing DNA ligase IV, XRCC4 and XRCC4-like factor (XLF; also
called Cernunnos or NHEJ1) (2,6,7). XLF
is structurally similar to XRCC4 and physically interacts with
XRCC4. It is recruited to the DSB site in a Ku-dependent manner at
an early stage of NHEJ (8,9) and facilitates XRCC4-mediated joining
of blunt ends and several types of mismatched ends, that are
non-complementary or partially complementary.
Deficiency in DDR pathways, including the NHEJ
pathway, lead to premature ageing (10). Ku70, Ku80 or double knockout mice
exhibit a series of age-related phenotypes, shortened life span and
accumulation of chromosomal instability (11). Increased spontaneous translocations
have also been identified in XLF-deficient murine embryonic
stem cells (12). However, the
link between XLF and ageing remains to be established.
Werner syndrome (WS) is a rare autosomal recessive
progeroid syndrome characterized by the premature onset of multiple
age-related disorders, including atherosclerosis, cancer
predisposition and premature ageing. The WRN gene, when
mutated, causes WS. The majority of the known WRN mutations
are nonsense, producing truncated proteins lacking the nuclear
localization signal (13). The WRN
protein possesses helicase (14)
and exonuclease (15) activities,
and is important in multiple DNA metabolism pathways including DNA
repair, recombination, replication and telomere maintenance
(16). During the process of NHEJ,
WRN is physically and functionally associated with, and regulated
by, the major NHEJ factors, including the Ku70/Ku80 heterodimer,
DNA-PKcs and the DNA ligase IV/XRCC4 complex, to optimize DNA
end-processing (17–21).
WRN also functions in DNA transcription. It promotes
RNA polymerase I-dependent transcription of ribosomal RNA (22) and is important in RNA polymerase II
(RNA pol II)-dependent transcription (23). Transcription alterations have been
identified in human fibroblasts from WS patients (24) and in the cells with RNAi-based
short-term knockdown of WRN (25). The efficiency of RNA pol II
transcription is reduced by ~50% in WRN-deficient cells
compared with that in normal cells. The gene expression profiles in
those cells resemble that of fibroblasts derived from old donor
patients (24,25). The transcription alterations in WS
are specific to sets of certain genes involved in different
biological pathways, including DNA repair, DNA replication and cell
cycle control (25), and may thus
contribute to the development of the WS phenotype.
In the present study, it was identified that XLF was
positively modulated by WRN and involved in the regulation of
cellular senescence.
Materials and methods
Cell lines, siRNA oligos and
antibodies
The human osteosarcoma cell line U2OS, human normal
fibroblast cell line WI38, human normal fibroblast cell line
GM00637 and human WRN-deficient fibroblast cell line AG11395
were purchased from the American Type Culture Collection (Manassas,
VA, USA). The cell lines were cultured in DMEM medium with 10%
fetal bovine serum (FBS; Hyclone Laboratories, Inc., Logan, UT,
USA) and grown at 37°C in the presence of 5% CO2.
All predesigned siRNA oligonucleotide duplexes
(OnTarget plus option) directed against human XLF or
WRN (si-XLF or si-WRN) were purchased from Dharmacon, Inc.
(Lafayette, CO, USA). The forward sequences of individual siRNA
oligonucleotide duplexes were as follows, for si1-XLF: GCA UUA CAG
UGC CAA GTG A dTdT; si2-XLF: CGC UGA UUC GAG AUC GAU UGA dTdT;
si1-WRN: CUG UAU CUU CGG GCA CCA A dTdT and i2-WRN: UGA AGA GCA AGU
UAC UUG C dTdT. The forward sequence of control siRNA
oligonucleotide duplex (si-CTR) was CGU ACG CGG AAU ACU UCG A
dTdT.
Mouse monoclonal antibody against β-actin (clone
AC15) was purchased from Sigma (St. Louis, MO, USA). Antibodies
against XLF (BL3263) and WRN (BL1309) were purchased from the
Bethyl Laboratories (Montgomery, TX, USA). Peroxidase-conjugated
secondary antibodies were from Jackson ImmunoResearch (West Grove,
PA, USA).
Cell growth assay
WI38 cells were transfected with si-XLF or si-CTR
with RNAiMAX. Cells were trypsinized 24 h following transfection
and transferred into 6-well plates (1×104 cells/well).
The cell number was counted every day for 5 days, with triplicated
wells being used at each time point.
Senescence-associated β-galactosidase
(SA-β-gal) staining
WI38 cells at passage 39 were infected with a
control siRNA (si-CTR) or XLF-specific siRNAs (si1-XLF or si2-XLF).
Transfectants were cultured for 10 days and processed for SA-β-gal
staining as described by Dimri et al (26). Briefly, the cells were washed with
PBS and fixed with 0.5% glutaraldehyde in PBS for 5 min at room
temperature. Following washing with PBS, the cells were incubated
with a freshly prepared staining solution [1 mg/ml
5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), 40 mmol/l
citric acid/sodium phosphate (pH 6.0), 5 mmol/l potassium
ferrocyanide, 5 mmol/l potassium ferricyanide, 15 mmol/l NaCl and 2
mmol/l MgCl2] at 37°C for 16 h.
Western blot analysis
Cell lysates were prepared in 0.5% NP-40 lysis
buffer [50 mmol/l Tris (pH 8.0), 250 mmol/l NaCl, 5 mmol/l EDTA,
0.5% NP40] containing protease inhibitor cocktail (Roche
Diagnostics, Indianapolis, IN, USA). The protein concentration was
determined using an DC assay kit (Bio-Rad, Hercules, CA, USA).
Equal amounts of proteins were resolved on 4–18% gradient SDS-PAGE
gels and transferred onto nitrocellulose membranes (Bio-Rad). The
blots on nitrocellulose were blocked with 5% non-fat milk in PBST
(PBS with 0.05% Tween-20) and were sequentially incubated with
primary antibodies as indicated and horseradish
peroxidase-conjugated secondary antibodies in 5% non-fat milk in
PBST. Blots were washed with PBST following each incubation. The
immunoreactive bands were visualized by Amersham Biosciences ECL
reagents ((Piscataway, NJ, USA) following the manufacturer’s
instructions.
Transfections and dual luciferase
reporter assays
siRNA oligonucleotide duplex at a final
concentration of 40 μM, was transfected into U2OS cells twice with
a 24 h interval using oligofectamine (Invitrogen Life Technologies,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
Transfectants were used for further experiments 24 h following the
secondary transfection. For dual luciferase reporter assays, on the
day prior to siRNA transfection, 5×104 cells were seeded
into each well of 24-well plates. Following the secondary siRNA
transfection (4 h), a firefly luciferase reporter construct under
the control of the XLF promoter (1 μg) and a Renilla
luciferase reporter construct under the control of the TK promoter
for normalization of transfection efficiency (10 ng) were
co-transfected into cells in triplicate using FuGENE6 (Roche
Diagnostics) at a ratio of 1 μg of plasmid to 3 μl of FuGENE6.
Luciferase activity was determined with the dual luciferase assay
system (Promega Corporation, Madison, WI) 48 h following the first
siRNA transfection. Relative light units were determined using a
luminometer (microtiter plate luminometer). Experiments were
performed at least three times independently and each combination
was tested in triplicate wells.
Reverse-transcription PCR and real-time
reverse-transcription PCR
Total RNA was extracted from U2OS cells using the
TRIzol reagent (Invitrogen Life Technologies). Reverse
transcription (RT)-PCR was performed using the Access Quick RT-PCR
system (Promega Corporation) essentially according to the
manufacturer’s instructions. Real-time PCR was performed using the
QuantiTect SYBR-Green PCR kit (Qiagen, Hilden, Germany) and the iQ5
thermal cycler (Bio-Rad). Primers used for amplification of GAPDH
were GAPDH: sense, TGG TAT CGT GGA AGG ACT CA and antisense, CCA
GAT GAG GCA GGG ATG AT. Primers used for amplification of XLF were
XLF: sense, GAG TCC ACG GGT ACT TCA GG and antisense, GGG CCT GTC
AAC ATC AAC TT.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as previously described
(27). The ChIP-enriched DNA was
amplified by PCR with primer pairs that are specific for the
XLF promoter sequence. XLF promoter first fragment
corresponds to the position from −596 to −337, primers used were
forward: GGT ACC GAA GGG ATA ATG AAT TCT GAT TGG GGA CAG and
reverse: GGA TCC TCC GAC CTC ATC CTT TAC CTC TCC TGC TTC.
XLF promoter second fragment corresponds to the position
from −360 to −90, primers used were forward: GGT ACC GGA GAG GTA
AAG GAT GAG GTC GGA CTA TG and reverse: GGA TCC GGC TAG TAG AAG GGT
AGT GGC GCG TCT TG. XLF promoter third fragment corresponds
to the position from −81 to +169, primers used were forward: GGT
ACC GGC CTC TCC TCC ACT TAC CCT GGC CAC TG and reverse: GGA TCC GAC
TCG AAC GCG ATT CCA CCT ACC GTC AG.
Results and Discussion
XLF is a transcriptional target of
WRN
A candidate gene approach was employed to identify
DDR factors in the human fibroblast cell line AG11395A, which was
originally isolated from a WRN patient bearing a nonsense
mutation in the WRN gene. It was identified that the
endogenous XLF protein level was lower in AG11395A cell line than
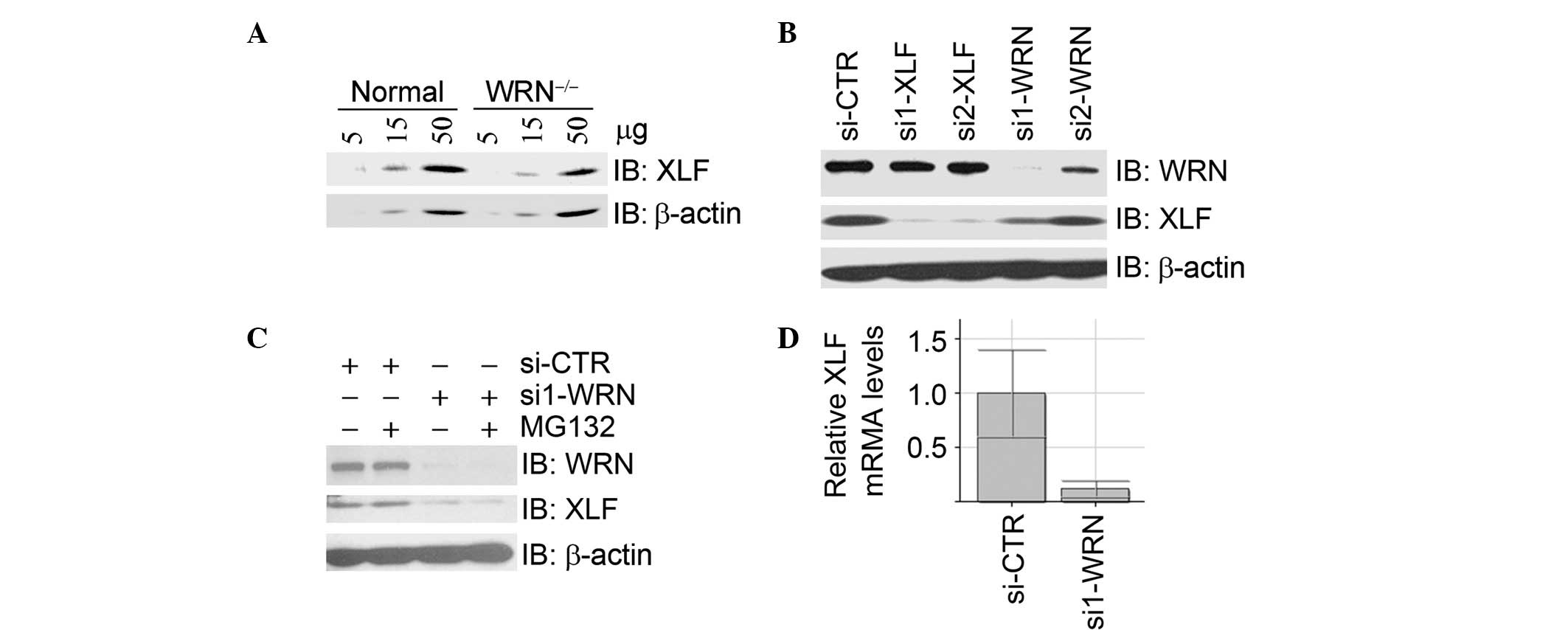
in the control fibroblast cell line GM00637G (Fig. 1A). In U2OS cells, inhibition of
WRN expression by two independent siRNA oligos resulted in a
decrease of XLF protein levels (Fig.
1B), which were not restored by treatment with the proteasome
inhibitor MG132 (Fig. 1C).
Real-time RT-PCR assays demonstrated that the mRNA levels of XLF
were significantly decreased upon depletion of WRN by siRNA
(Fig. 1D). Taken together, these
data suggest that WRN is a positive regulator of XLF at the
transcriptional level.
WRN positively regulates the XLF promoter
activity
To determine if WRN regulates the promoter activity
of XLF, the putative XLF promoter region from −596 to +169
(relative to the putative transcription start site described in the
Ensembl protein-coding gene ENSG00000187736) was cloned into the
pGL3 basic luciferase reporter vector. The resulting pGL3-XLFpr was
identified to have pronounced luciferase reporter activity in U2OS
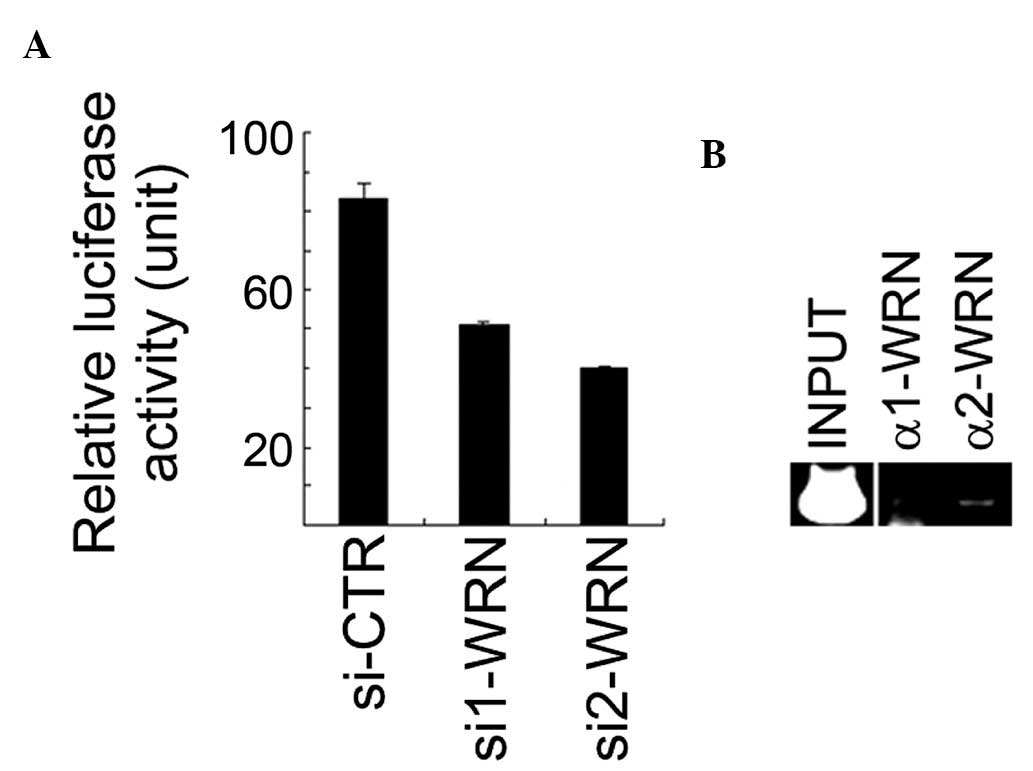
cells (data not shown). It was identified that the XLF
promoter activity was significantly downregulated when WRN
expression was inhibited by siRNA in U2OS cells (Fig. 2A). The antibodies against WRN were
used for ChIP on cross-linked chromatin fragments prepared from
U2OS cells. The ChIP-enriched DNA was subjected to PCR analysis
using three pairs of primers for amplification of three fragments
within the XLF promoter region. The data revealed that the
second fragment (from −360 to −90) of the XLF promoter
region was detected in the anti-WRN immunoprecipitates (Fig. 2B). These results suggest that WRN
resides on the XLF promoter region, positively regulating
its activity.
Depletion of XLF promotes cellular
senescence in normal human fibroblasts
The prominent biological function of XLF is to
repair DSBs by NHEJ, while the central effect of WRN
deficiency is premature cellular senescence. It is well known that
defects in NHEJ-mediated DSB repair contribute to cellular
senescence. Therefore, we hypothesized that defects in XLF
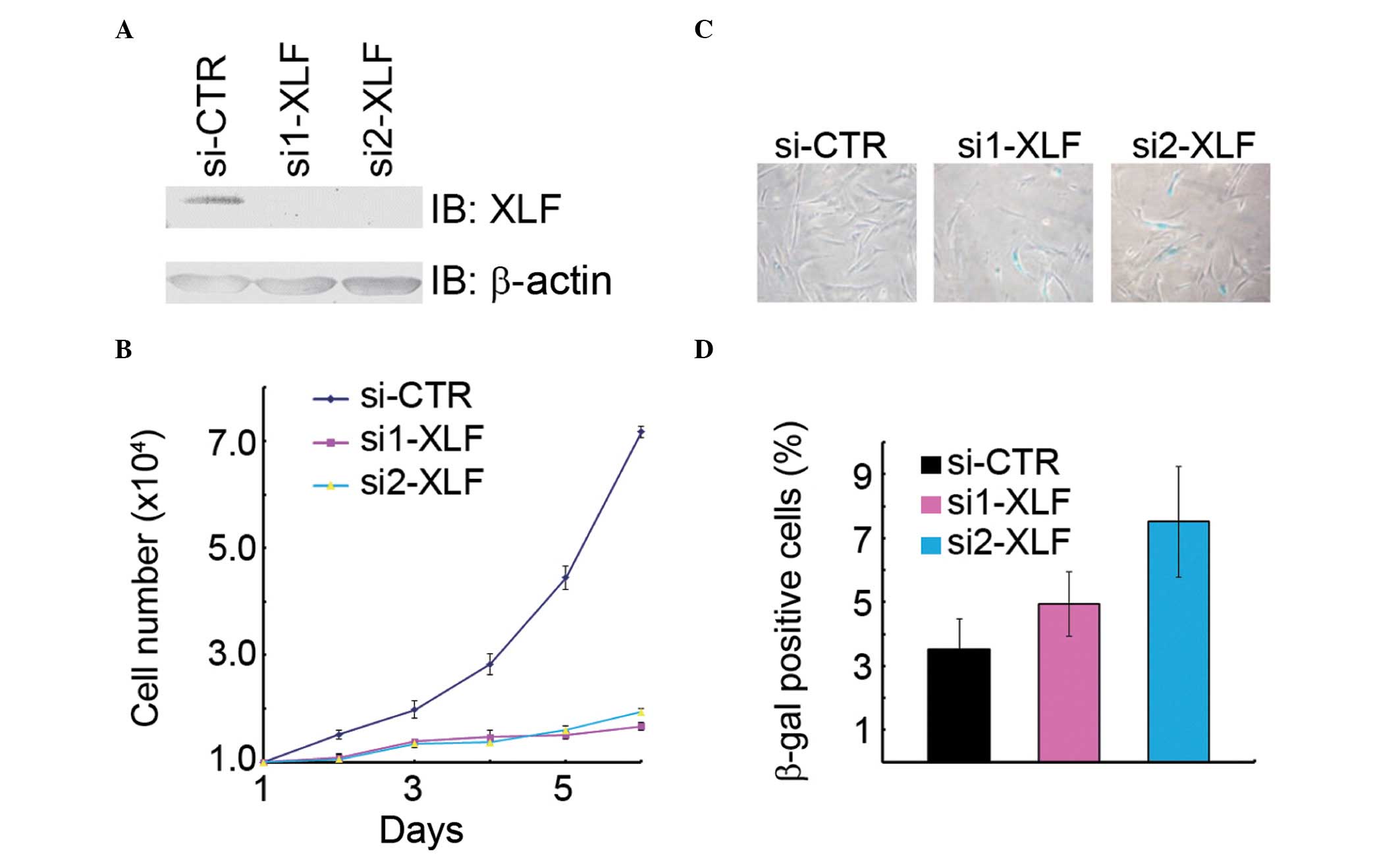
would lead to premature senescence. Indeed, inhibition of
XLF expression by transfection with two independent siRNA
oligos resulted in a decrease of cell growth in the normal human
fibroblasts WI38 (Fig. 3A and B),
while an increase in the percentage of β-gal-positive cells as
compared with the mock transfectants (Fig. 3C). Therefore, it was concluded that
XLF is critical in promoting cell proliferation and suppressing
cellular senescence.
WRN is known to physically and functionally interact
with the DNA-PK kinase complex and the XRCC4/Ligase IV complex, two
key complexes involved in NHEJ. The DNA-PK kinase complex has been
demonstrated to phosphorylate WRN in vivo and in
vitro, and this phosphorylation inhibits the WRN helicase and
exonuclease activities. By contrast, the interaction between XRCC4
and WRN, stimulates WRN’s exonuclease activity, enabling it to
serve as a DNA end-processing factor during NHEJ. In the present
study, the results reveal a novel mechanism of the functional
interplay between WRN and the NHEJ process, in which WRN positively
regulates XLF at the transcription level. Furthermore, these data
provide evidence for the first time, to the best of our knowledge,
of the functional link of XLF to cellular senescence.
Acknowledgements
We thank Eric W. McIntush from the Bethyl
Laboratories for antibodies against XLF and its interacting
proteins. We thank other members of the Xu laboratory for help.
This study was supported by the National Natural Science Foundation
of China (31130017 and 31071190), the 973 project 2013CB911002,
Research Fund for the Doctoral Program of Higher Education of China
(20101108110002) and Funding Project for Academic Human Resources
Development in Institutions of Higher Learning under the
Jurisdiction of Beijing Municipality (PHR20110508) to X.X. and the
973 project 2012CB911203 to YSC.
References
|
1
|
Jackson SP: Sensing and repairing DNA
double-strand breaks. Carcinogenesis. 23:687–696. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ciccia A and Elledge SJ: The DNA damage
response: making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lees-Miller SP and Meek K: Repair of DNA
double strand breaks by non-homologous end joining. Biochimie.
85:1161–1173. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mladenov E and Iliakis G: Induction and
repair of DNA double strand breaks: the increasing spectrum of
non-homologous end joining pathways. Mutat Res. 711:61–72. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Buck D, Malivert L, de Chasseval R, et al:
Cernunnos, a novel nonhomologous end-joining factor, is mutated in
human immunodeficiency with microcephaly. Cell. 124:287–299. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ahnesorg P, Smith P and Jackson SP: XLF
interacts with the XRCC4-DNA ligase IV complex to promote DNA
nonhomologous end-joining. Cell. 124:301–313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yano K, Morotomi-Yano K, Wang SY, et al:
Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 9:91–96.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yano K and Chen DJ: Live cell imaging of
XLF and XRCC4 reveals a novel view of protein assembly in the
non-homologous end-joining pathway. Cell Cycle. 7:1321–1325. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hasty P: Is NHEJ a tumor suppressor or an
aging suppressor? Cell Cycle. 7:1139–1145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li H, Vogel H, Holcomb VB, Gu Y and Hasty
P: Deletion of Ku70, Ku80, or both causes early aging without
substantially increased cancer. Mol Cell Biol. 27:8205–8214. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zha S, Alt FW, Cheng HL, Brush JW and Li
G: Defective DNA repair and increased genomic instability in
Cernunnos-XLF-deficient murine ES cells. Proc Natl Acad Sci USA.
104:4518–4523. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martin GM, Austad SN and Johnson TE:
Genetic analysis of ageing: role of oxidative damage and
environmental stresses. Nat Genet. 13:25–34. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gray MD, Shen JC, Kamath-Loeb AS, et al:
The Werner syndrome protein is a DNA helicase. Nat Genet.
17:100–103. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang S, Li B, Gray MD, Oshima J, Mian IS
and Campisi J: The premature ageing syndrome protein, WRN, is a
3′-->5′ exonuclease. Nat Genet. 20:114–116. 1998.
|
|
16
|
Luo J: WRN protein and Werner syndrome. N
Am J Med Sci (Boston). 3:205–207. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li B and Comai L: Functional interaction
between Ku and the werner syndrome protein in DNA end processing. J
Biol Chem. 275:398002000.PubMed/NCBI
|
|
18
|
Yannone SM, Roy S, Chan DW, et al: Werner
syndrome protein is regulated and phosphorylated by DNA-dependent
protein kinase. J Biol Chem. 276:38242–38248. 2001.PubMed/NCBI
|
|
19
|
Li B and Comai L: Displacement of DNA-PKcs
from DNA ends by the Werner syndrome protein. Nucleic Acids Res.
30:3653–3661. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Otsuki M, Seki M, Kawabe Y, et al: WRN
counteracts the NHEJ pathway upon camptothecin exposure. Biochem
Biophys Res Commun. 355:477–482. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kusumoto R, Dawut L, Marchetti C, et al:
Werner protein cooperates with the XRCC4-DNA ligase IV complex in
end-processing. Biochemistry. 47:7548–7556. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shiratori M, Suzuki T, Itoh C, Goto M,
Furuichi Y and Matsumoto T: WRN helicase accelerates the
transcription of ribosomal RNA as a component of an RNA polymerase
I-associated complex. Oncogene. 21:2447–2454. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Balajee AS, Machwe A, May A, et al: The
Werner syndrome protein is involved in RNA polymerase II
transcription. Mol Biol Cell. 10:2655–2668. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kyng KJ, May A, Kølvraa S and Bohr VA:
Gene expression profiling in Werner syndrome closely resembles that
of normal aging. Proc Natl Acad Sci USA. 100:12259–12264. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Turaga RV, Paquet ER, Sild M, et al: The
Werner syndrome protein affects the expression of genes involved in
adipogenesis and inflammation in addition to cell cycle and DNA
damage responses. Cell Cycle. 8:2080–2092. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dimri GP, Lee X, Basile G, et al: A
biomarker that identifies senescent human cells in culture and in
aging skin in vivo. Proc Natl Acad Sci USA. 92:9363–9367. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rauch T, Zhong X, Pfeifer GP and Xu X:
53BP1 is a positive regulator of the BRCA1 promoter. Cell Cycle.
4:1078–1083. 2005. View Article : Google Scholar : PubMed/NCBI
|