Introduction
Repolarization of the cardiac action potential is
accomplished by several types of potassium currents. One of these,
the rapid component of delayed rectifier potassium current
(IKr), is unique in its ability to modify membrane
repolarization at the end of each cardiac action potential
(1). Activation of IKr,
which is predominantly carried through the human
ether-à-go-go-related gene (hERG) potassium ion channels, initiates
membrane repolarization and terminates the plateau phase of the
cardiac action potential (2).
Mutation in hERG or pharmacological blockade of the hERG channels
can produce an excessive prolongation of the action potential
duration and the QT interval, leading to proarrhythmic events
usually characterized by polymorphic ventricular tachycardia or
torsades de pointes (3–9). Such cardiac electrical disturbances
are often closely correlated with physical or emotional stress,
particularly in patients with hereditary long QT syndrome,
indicating a potential correlation between adrenergic stimulation
and hERG potassium channel activity (10).
Previous studies have revealed that
hERG/IKr currents are modulated by α- and β-adrenergic
stimulation, thus providing a pathophysiological rationale for an
increased incidence of arrhythmias during stress (11–14).
In human hearts, there are several main subfamilies of the
adrenergic receptor (adrenoceptor) family, namely α1-,
α2-, β1-, β2- and
β3-adrenoceptors. Our previous study found that
IKr currents in the guinea-pig left ventricular myocytes
was regulated by α1-adrenergic stimulation via protein
kinase C (PKC)- and protein kinase A (PKA)-dependent pathways
(15).
The β1- and β2-adrenoceptors
are the predominant subtypes in the heart. In human myocardium,
β1-adrenoceptors constitute 70–80% of the total
β-adrenoceptors abundance (16)
and an altered β1-adrenoceptor activity and/or signaling
are associated with a high incidence of cardiac arrhythmias
(17). β1-adrenoceptor
coupled with Gs-protein stimulates adenylate cyclase (AC),
resulting in the accumulation of cyclic adenosine monophosphate
(AMP) and the activation of PKA. The activation of the AC/cAMP/PKA
pathway results in a complex regulation of hERG/IKr.
However, whether PKC and PLC are involved in
β1-adrenoceptor-induced regulation of IKr
remains unclear. The present study aimed to investigate how
IKr is regulated in guinea-pig cardiomyocytes following
activation of β1-adrenergic receptors, and the
involvement of activation of PKA, PKC and PLC.
Materials and methods
Animal and myocyte isolation
All experiments were approved by Animal Care
Protocols of Nanjing Medical University Institutional Animal Care
and Use Committee (Nanjing, China). Single left ventricular
myocytes were enzymatically isolated from guinea-pig heart as
described previously (18) with
minor modifications. Briefly, male healthy guinea pigs (weight,
300–350 g; provided by the Experimental Animal Center of Jiangsu
Province, China) were sacrificed by cervical dislocation, and the
heart was then rapidly removed and cannulated at the aorta.
Following perfusion with an enzymatic solution, the left
ventricular tissue was excised from the softened hearts, minced,
and simultaneously filtered cardiomyocytes were stored at 4°C prior
to patch clamp recording.
Electrophysiology recording
Cardiomyocytes were transferred to a recording
chamber (Warner TC-324B; Warner Instruments, Hamden, CT, USA)
continuously perfused with the bath solution. Pipettes had
resistances of 3–6 MΩ subsequent to filling with the pipette
solution. Whole-cell patch-clamp recordings were performed with an
EPC-9 amplifier (HEKA, Lambrecht, Germany). All the recordings were
conducted at 37±0.5°C and the flow rate was maintained at ~2 ml
min−1.
Solutions and drugs
In order to record the IKr current, the
pipette solution contained 140 mmol l−1 KCl, 1 mmol
l−1 CaCl2, 2 mmol l−1
MgCl2, 10 mmol l−1 HEPES, 11 mmol
l−1 EGTA, 5 mmol l−1 Na2-ATP and 5
mmol l−1 creatine phosphate (disodium salt); pH 7. 4
adjusted with 8 M KOH. The bath solution contained 140 mmol
l−1 NaCl, 3.5 mmol l−1 KCl, 1.5 mmol
l−1 CaCl2, 1.4 mmol l−1
MgSO4 and 10 mmol l−1 HEPES; pH adjusted to
7.4 with 10 M NaOH. Calcium currents were blocked by 10 μM
nifedipine in the bath solution and 10 μM chromanol 293B was used
to ablate the slow component of the delayed rectifier potassium
currents (IKs). Na2-ATP, EGTA, nifedipine,
chromanol 293B, chelerythrine, U73122 and xamoterol were purchased
from Sigma (Sigma-Aldrich, St. Louis, MO, USA), collagenase II from
Worthington (Lakewood, NJ, USA) and KT5720 from Merck (Darmstadt,
Germany). Dofetilide, a specific blocker of IKr or hERG,
was provided by Pfizer (Shanghai, China). All the other reagents
were purchased from Amresco (Solon, OH, USA).
For stock solutions, dofetilide was dissolved in
distilled water to a concentration of 10 mM; KT5720, chelerythrine
and U73122 were dissolved in dimethylsulfoxide (DMSO) to a
concentration of 2.5 mM, 1 mM and 0.1 mM. These chemicals were
stored at −20°C until further use. The final concentration of DMSO
was <0.5% in the bath solution and exerted no effect on the
currents that were observed.
Quantification and statistics
Following initiation of the test pulse, tail
currents were measured. Changes in the current amplitude were
normalized prior to the application of xamoterol. All the data were
acquired by Pulse + Pulsefit V8.53 (HEKA Elektronik, Lambrecht,
Germany) and were analyzed by SPSS 18.0 software (SPSS Inc.,
Chicago, IL, USA). Statistical data were presented as the mean ±
standard error of the mean. A paired-sample t-test was used for
determining significant differences prior to and following the
xamoterol intervention. One-way analysis of variance, with a post
hoc comparison using a Newman-Keuls test was performed to compare
the differences among groups. P<0.05 was considered to indicate
a statistically significant difference.
Results
Effects of xamoterol on IKr
tail currents
A representative IKr tail current from a
guinea-pig ventricular myocyte is shown in Fig. 1A. In Fig. 1B, the current was completely
blocked by 1 μM dofetilide, a specific inhibitor of IKr,
indicating lack of contribution from any other current to the tail
current in the experimental settings of the present study. The
dose-dependent effects of xamoterol, a specific
β1-adrenoceptor agonist, on IKr current
amplitude were examined in freshly isolated guinea-pig ventricular
myocytes.
Fig. 2A shows a
representative current trace when the cardiomyocyte was treated
with by 0.01 μM-100 μM xamoterol. Fig.
2B shows the concentration-dependent reduction of
IKr elicited by xamoterol in cardiomyocytes. In the
cardiomyocytes examined (n=5), the bath application of 0.01, 0.1,
1, 10 and 100 μM xamoterol significantly reduced the IKr
current amplitude to 0.96±0.12, 0.86±0.13, 0.67±0.12, 0.59±0.10 and
0.55±0.11 respectively, compared with the basic amplitude. The
concentration of 10 μM was selected for xamoterol for the rest of
this study, since this concentration had almost decreased the
current by the maximum degree. To examine whether the
xamoterol-induced effect was β1-adrenoreceptor-mediated,
the specific β1-adrenoceptor blocker 10 μM atenolol was
coincubated with 10 μM xamoterol. This resulted in a decrease in
the current amplitude to only 0.87±0.05 at +40 mV, significantly
different from the current treated with 10 μM xamoterol alone
(0.56±0.04) (Fig. 3). These
results indicate that IKr is regulated by
β1-adrenoceptors in guinea-pig cardiomyocytes.
Effects of PKA inhibitor on
xamoterol-induced inhibition of IKr
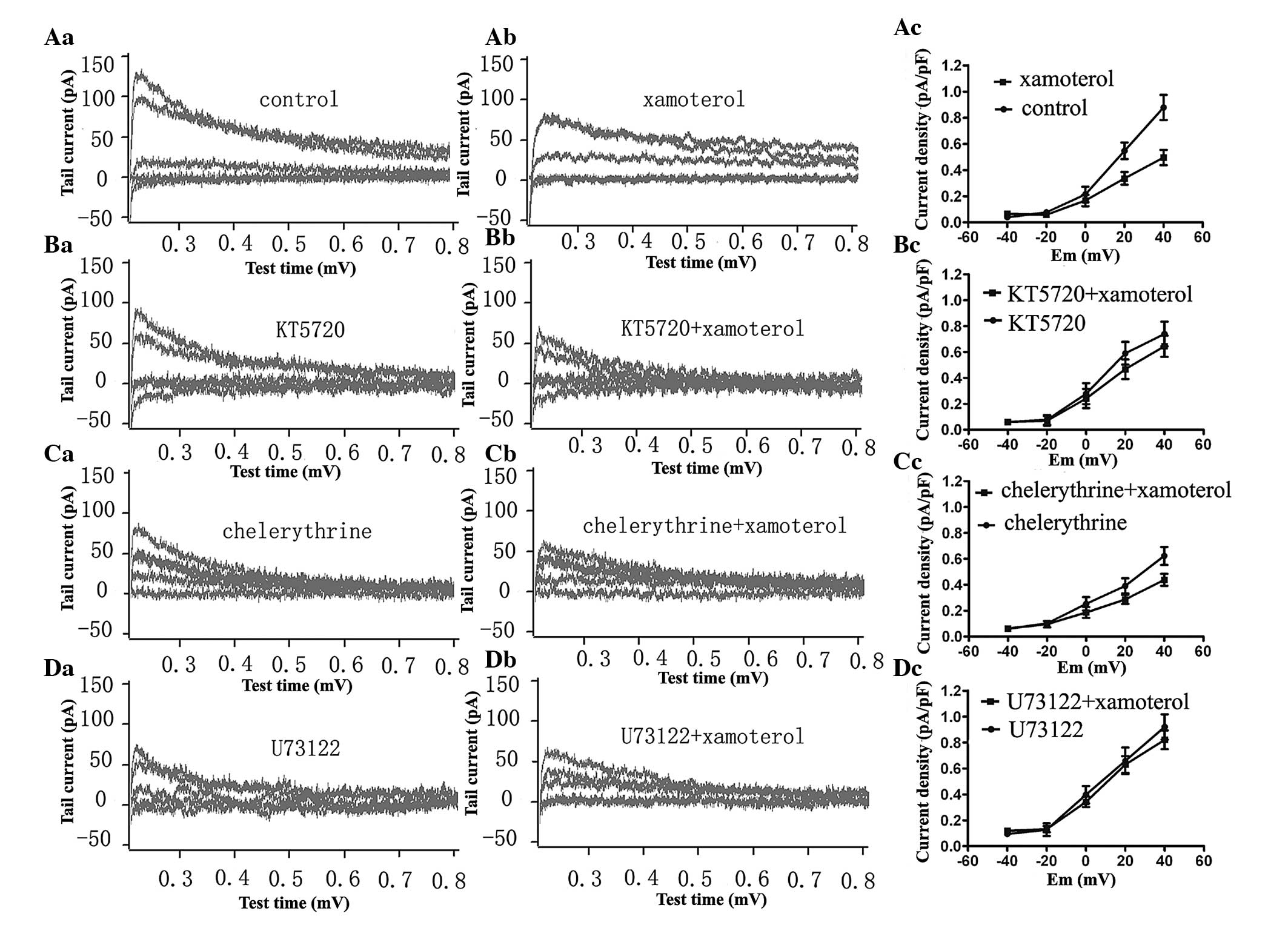
Cardiomyocytes were pretreated with 2.5 μM KT5720, a
specific PKA inhibitor, for 1 h prior to the examination of the 10
μM xamoterol-elicited effect on IKr. The IKr
tail current and the current density-voltage curve prior to and
following administration of xamoterol in cells pretreated with
KT5720 is shown in Fig. 4B. In the
present study, the IKr tail current density decreased
from 0.74±0.09 to 0.64±0.08 pA/pF at +40 mV. However, 10 μM
xamoterol was found to reduce the IKr tail current
density from 0.88±0.09 to 0.50±0.05 pA/pF at +40 mV in the presence
of an empty bath solution (Fig.
4A). In other words, the IKr tail current amplitude
was reduced to 0.87±0.03 subsequent to 10 μM xamoterol in the
presence of KT5720 (the second column in Fig. 5), which was significantly different
from that in the absence of KT5720, 0.56±0.04 (the control group,
the first column in Fig. 5). These
data demonstrate that a xamoterol-induced decrease in
IKr was reversed by KT5720.
Xamoterol-induced inhibition of
IKr is antagonized by the PKC inhibitor chelerythrine
and the PLC inhibitor U73122
The guinea-pig left ventricular myocytes were
pretreated with the 1 μM specific PKC inhibitor chelerythrine or
100 nM PLC inhibitor U73122 for one hour, and then the
IKr tail currents prior to and following xamoterol
administration were examined. Xamoterol reduced the IKr
tail current density from 0.62±0.07 to 0.44±0.05 pA/pF at +40 mV
(Fig. 4C), and it decreased
IKr to 0.71±0.01 pA/pF in the presence of chelerythrine
(Fig. 5), which was significantly
different from that in the absence of chelerythrine, the control
group, 0.56±0.04 pA/pF (Fig. 5).
The current trace and the tail current density-voltage (Id-V) curve
almost superimposed prior to and following xamoterol treatment in
the presence of U73122 (Fig. 4D),
from 0.92±0.09 to 0.82±0.07 pA/pF at +40 mV, indicating that
xamoterol failed to suppress IKr when myocytes were
pretreated with the PLC inhibitor. However, the effects of
xamoterol were significantly different between the control and the
U73122 group (Fig. 5).
Discussion
The present study indicated that xamoterol inhibits
IKr through β-adrenoceptors in freshly isolated
guinea-pig cardiomyocytes. Furthermore, this inhibitory effect was
significantly attenuated by the PKA inhibitor KT5720, the PLC
inhibitor U73122 and the PKC inhibitor chelerythrine. These data
indicated the involvement of PKA, PKC and PLC activation in the
β1-adrenoceptor-induced inhibition of the IKr
current.
Activation of β1-adrenoceptors has been
demonstrated to elicit an inhibitory effect on IKr or
hERG via a cAMP/PKA-dependent pathway (11,19)
consistent with our data that the PKA inhibitor KT5720 attenuated
the inhibitory effect of xamoterol. However, in an early report by
Heath and Terrar (20), a
concentration-independent increase of the IKr currents
was observed at low concentrations of the β1-adrenergic
agonist isoprenaline and the stimulatory effect of isoprenaline on
IKr was inhibited by the selective PKC inhibitor
bisindolylmaleimide I. There may be several reasons for the
disparate findings between the two laboratories, including
differences in patch-clamp modes and experimental conditions,
aswell as dual regulation of hERG by cAMP and PKA phosphorylation
(21,22). Nonetheless, to the best of our
knowledge the involvement of PKC in β1-adrenergic
regulation of IKr demonstrated in the present study has
not been documented previously.
PKC is an important member of the signaling
transduction pathway, capable of reducing hERG currents through a
mechanism independent of PKC-elicited phosphorylation of hERG
(23,24). The results of the present study
indicated that xamoterol-induced inhibition of IKr is
partially modulated by PKC. In addition, the decrease in
IKr induced by xamoterol was also antagonized by the
selective PLC inhibitor U73122. Usually PLC is linked to
α1-adrenergic stimulation leading to the PIP2
hydrolysis and then the activation of PKC. PIP2
depletion has been shown to alter the cardiac IKr
current (25,26). PKC has also been shown to reduce
the hERG current (27). Although
PKC and PLC are associated with the classical
α1-adrenergic signaling pathway, the results of the
present study reveal that PKC and PLC are also activated in the
β1-adrenergic signaling pathway in the regulation of the
IKr/hERG currents, indicating that there may exist a
‘cross-talk’ between the α1- and
β1-adrenergic signaling cascades. Therefore, our next
aim is to analyze the details in this type of cross-talk and
demonstrate direct evidence.
In conclusion, the present study demonstrates that
IKr is regulated by β1-adrenergic receptors
in guinea-pig cardiomyocytes, via the PKA-, PKC- and/or
PLC-dependent signaling pathways. These findings provide a possible
correlation between stress and life-threatening arrhythmias and may
provide insight into the pathogenesis and potential therapeutic
strategies for clinical cardiac arrhythmias.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81100123) and the
Postgraduate Innovation Projects of Jiangsu Province (grant no.
JX22013176). The authors would like to acknowledge the helpful
support from Professor Di Yang, Professor Xiang-Jian Chen and
Professor Hen-Fang Wu (Department of Cardiology, First Affiliated
Hospital of Nanjing Medical University).
References
|
1
|
Thomas D, Karle CA and Kiehn J: The
cardiac hERG/IKr potassium channel as pharmacological target:
structure, function, regulation, and clinical applications. Curr
Pharm Des. 12:2271–2283. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Charpentier F, Mérot J, Loussouarn G and
Baró I: Delayed rectifier K(+) currents and cardiac repolarization.
J Mol Cell Cardiol. 48:37–44. 2010.
|
|
3
|
Sanguinetti MC, Jiang C, Curran ME and
Keating MT: A mechanistic link between an inherited and an acquired
cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell.
81:299–307. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ficker E, Thomas D, Viswanathan PC, et al:
Novel characteristics of a misprocessed mutant HERG channel linked
to hereditary long QT syndrome. Am J Physiol Heart Circ Physiol.
279:H1748–H1756. 2000.PubMed/NCBI
|
|
5
|
Thomas D, Gut B, Wendt-Nordahl G and Kiehn
J: The antidepressant drug fluoxetine is an inhibitor of human
ether-a-go-go-related gene (HERG) potassium channels. J Pharmacol
Exp Ther. 300:543–548. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roden DM: Human genomics and its impact on
arrhythmias. Trends Cardiovasc Med. 14:112–116. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du L, Li M, You Q and Xia L: A novel
structure-based virtual screening model for the hERG channel
blockers. Biochem Biophys Res Commun. 355:889–894. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Noord C, Sturkenboom MC, Straus SM,
Witteman JC and Stricker BH: Non-cardiovascular drugs that inhibit
hERG-encoded potassium channels and risk of sudden cardiac death.
Heart. 97:215–220. 2011.PubMed/NCBI
|
|
9
|
Vandenberg JI, Perry MD, Perrin MJ, Mann
SA and Hill AP: hERG K(+) channels: structure, function, and
clincal signifcance. Physiol Rev. 92:1393–1478. 2012.
|
|
10
|
Tutor AS, Delpón E, Caballero R, et al:
Association of 14-3-3 proteins to beta1-adrenergic receptors
modulates Kv11.1 K+ channel activity in recombinant
systems. Mol Biol Cell. 17:4666–4674. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Karle CA, Zitron E, Zhang W, Kathöfer S,
Schoels W and Kiehn J: Rapid component I(Kr) of the guinea-pig
cardiac delayed rectifier K(+) current is inhibited by
beta(1)-adrenoreceptor activation, via cAMP/protein kinase
A-dependent pathways. Cardiovasc Res. 53:355–62. 2002.
|
|
12
|
Thomas D, Kiehn J, Katus HA and Karle CA:
Adrenergic regulation of the rapid component of the cardiac delayed
rectifier potassium current, I(Kr), and the underlying hERG ion
channel. Basic Res Cardiol. 99:279–287. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zankov DP, Yoshida H, Tsuji K, Toyoda F,
Ding WG, Matsuura H and Horie M: Adrenergic regulation of the rapid
component of delayed rectifier K+ current: implications for
arrhythmogenesis in LQT2 patients. Heart Rhythm. 6:1038–1046.
2009.
|
|
14
|
Chen J, Chen K, Sroubek J, Wu ZY, Thomas
D, Bian JS and McDonald TV: Post-transcriptional control of human
ether-a-go-go-related gene potassium channel protein by
alpha-adrenergic receptor stimulation. Mol Pharmacol. 78:186–197.
2010. View Article : Google Scholar
|
|
15
|
Wang S, Xu DJ, Cai JB, Huang YZ, Zou JG
and Cao KJ: Rapid component I(Kr) of cardiac delayed rectifier
potassium currents in guinea-pig is inhibited by
alpha(1)-adrenoreceptor activation via protein kinase A and protein
kinase C-dependent pathways. Eur J Pharmacol. 608:1–6. 2009.
View Article : Google Scholar
|
|
16
|
Brodde OE and Michel MC: Adrenergic and
muscarinic receptors in the human heart. Pharmacol Rev. 51:651–690.
1999.PubMed/NCBI
|
|
17
|
Olson EN: A decade of discoveries in
cardiac biology. Nat Med. 10:467–474. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong DL, Liu Y, Zhou YH, Song WH, Wang H
and Yang BF: Decreases of voltage-dependent K+ currents
densities in ventricular myocytes of guinea pigs by chronic oxidant
stress. Acta Pharmacol Sin. 25:751–755. 2004.PubMed/NCBI
|
|
19
|
Li Y, Sroubek J, Krishnan Y and McDonald
TV: A-kinase anchoring protein targeting of protein kinase A and
regulation of HERG channels. J Membr Biol. 223:107–116. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heath BM and Terrar DA: Protein kinase C
enhances the rapidly activating delayed rectifier potassium
current, IKr, through a reduction in C-type inactivation in
guinea-pig ventricular myocytes. J Physiol. 522:391–402. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui J, Melman Y, Palma E, Fishman GI and
McDonald TV: Cyclic AMP regulates the HERG K(+) channel by dual
pathways. Curr Biol. 10:671–674. 2000.
|
|
22
|
Zankov DP, Yoshida H, Tsuji K, Toyoda F,
Ding WG, Matsuura H and Horie M: Adrenergic regulation of the rapid
component of delayed rectifier K+ current: implications
for arrhythmogenesis in LQT2 patients. Heart Rhythm. 6:1038–1046.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kiehn J, Karle C, Thomas D, Yao X,
Brachmann J and Kübler W: HERG potassium channel activation is
shifted by phorbol esters via protein kinase A-dependent pathways.
J Biol Chem. 273:25285–25291. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thomas D, Kiehn J, Katus HA and Karle CA:
Defective protein trafficking in hERG-associated hereditary long QT
syndrome (LQT2): molecular mechanisms and restoration of
intracellular protein processing. Cardiovasc Res. 60:235–241. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bian JS, Kagan A and McDonald TV:
Molecular analysis of PIP2 regulation of HERG and IKr. Am J Physiol
Heart Circ Physiol. 287:H2154–H2163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bian JS and McDonald TV:
Phosphatidylinositol 4,5-bisphosphate interactions with the HERG
K(+) channel. Pflugers Arch. 455:105–113. 2007.
|
|
27
|
Cockerill SL, Tobin AB, Torrecilla I,
Willars GB, Standen NB and Mitcheson JS: Modulation of hERG
potassium currents in HEK-293 cells by protein kinase C. Evidence
for direct phosphorylation of pore forming subunits. J Physiol.
581:479–493. 2007. View Article : Google Scholar : PubMed/NCBI
|