Introduction
Apoptosis is a key event in the progression of
advanced atherosclerosis. Macrophage apoptosis occurs throughout
all stages of atherosclerosis. Research directed at understanding
the consequences of macrophage death in atherosclerosis has
revealed opposing roles of apoptosis in atherosclerotic plaque
progression (1). Apoptosis of
macrophages is particularly important in plaque development and
breakdown and may be induced by uptake of oxidized low-density
lipoprotein (ox-LDL) or by loading with free cholesterol (FC)
(2). Increasing evidence suggests
that elevation of cellular cholesterol has a critical role in the
regulation of ox-LDL-mediated apoptosis, and factors regulating the
accumulation or elimination of lipids from macrophages are critical
in the formation of early atherosclerotic lesions and progression
to the chronic stage of atherosclerosis (3–6).
Thus, the acceleration of cholesterol efflux or prevention of the
accumulation of excess cholesterol in cells may inhibit
ox-LDL-induced apoptosis, which may provide new strategies for the
prevention and reversal of atherosclerosis.
A dual-domain peptide has been designed that
possesses the 141- to 150-residue arginine-rich domain
(LRKLRKRLLR), of apolipoprotein (apo) E, covalently linked to the
well-characterized class A amphipathic helical peptide 18A, a
high-affinity lipid-associating peptide (DWLKAFYDKVA-EKLKEAF)
(7). The resulting peptide
(Ac-hE-18A-NH2) has been shown to promote the rapid
uptake and clearance of atherogenic apo B-containing lipoproteins
in vitro and in dyslipidemic mouse models (8–10).
Furthermore, in the Watanabe heritable hyperlipidemic rabbit, a
single administration of the peptide Ac-hE-18A-NH2 not
only reduced plasma cholesterol levels, but also restored
endothelial function (11–12).
The association of peptide Ac-hE-18A-NH2
to apo B-containing lipoproteins is a prerequisite for the
clearance of these lipoproteins (10). Certain class A peptides have been
shown to remove ‘seeding molecules’ that are oxidized products of
arachidonic and linoleic acids from the LDL surface (13–14).
As the peptide Ac-hE-18A-NH2 possesses a class A
amphipathic helical domain that has been shown to possess several
apo A-I-mimetic properties, it was hypothesized in the present
study that this peptide, in addition to significantly reducing
plasma cholesterol levels, was also likely to exert a protective
effect against ox-LDL-induced apoptosis of macrophages, and that
this effect was likely to be associated with cholesterol
efflux.
Materials and methods
Peptide synthesis
The peptide Ac-hE-18A-NH2 with the
sequence Ac-LRKLRKRLLR-DWLKAFYDKVAEKLKEAF-NH2 was
synthesized using 9-fluorenylmethoxycarbonyl chemistry in an
automatic peptide synthesizer (PE Biosystems, Foster City, CA, USA)
according to the previously described procedure (9). The purity of the synthetic peptides
(typically >98%) was established by analytical high-performance
liquid chromatography and confirmed by mass spectral analysis.
Cell culture
RAW264.7 (Xiangya Medical College, Changsha, China),
a murine macrophage cell line, was maintained in Dulbecco’s
modified Eagle’s medium (DMEM, high glucose) supplemented with 10%
fetal bovine serum (Gibco; Invitrogen, Carlsbad, CA, USA) and 1%
antibiotics (penicillin and streptomycin). Cells were grown at 37°C
and 5% CO2 in humidified air. To further investigate the
role of cholesterol efflux on apoptosis induced by ox-LDL, in
certain experiments, the cells were co-treated with
Ac-hE-18A-NH2 and β-cyclodextrin (a cholesterol efflux
stimulator) or BFA (a cholesterol efflux blocker) for 24 h.
Cell viability assay
Following pre-incubation of the RAW264.7 cells
(1×105 cells/ml) in DMEM for 24 h, the peptide
Ac-hE-18A-NH2 (0–100 μg/ml) was added to the cells prior
to incubation for 24 and 48 h, respectively. The cytotoxic effect
of the peptide was then evaluated using the conventional MTT assay.
A total of 25 μl MTT (Sigma, St. Louis, MO, USA) solution [5 mg/ml
in phosphate-buffered saline (PBS), pH 7.4] was added and the cells
were incubated for 4 h. The incubation was terminated by addition
of 150 μl dimethylsulfoxide (Sigma). The absorbance at 492 nm was
assessed using a Varioskan Flash (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA).
Flow cytometric assessment of
apoptosis
Cultured RAW264.7 cells were collected and washed
twice with PBS. The measurement of phosphatidylserine
redistribution in a plasma membrane was conducted according to the
manufacturer’s instructions for the Annexin V-FITC Apoptosis
Detection kit (FITC, fluorescein isothiocyanide; Bender MedSystems,
Vienna, Austria). Cells were resuspended in binding buffer (1X),
and the cell density was adjusted to 1–10×106/ml, prior
to addition of 5 μl annexin V-FITC and 10 μl propidium iodide (PI;
20 μg/ml) to 100 μl cell suspension. The suspension was agitated
and incubated for 15 min at room temperature, under exclusion of
light. Following the addition of 400 μl binding buffer (1X),
fluorescence-activated cell sorting (FACS) analysis was performed
using the flow cytometer (Becton Dickinson, Franklin, NJ, USA).
Annexin V+/PI− cells were considered to be
apoptotic cells.
Caspase-3 activity assay
The activity of caspase-3 was determined using the
Caspase-3 Activity Assay kit (Beyotime Institute of Biotechnology,
Haimen, China). To evaluate the activity of caspase-3, cell lysates
were prepared following their respective incubation with various
designated agents. Assays were performed in 96-well microtiter
plates by incubating 10 μl cell lysate per sample in 80 μl reaction
buffer containing 10 μl caspase-3 substrate (Ac-DEVD-pNA, 2 mM).
Lysates were incubated at 37°C for 4 h. The concentration of the
p-nitroanilide (pNA) released from the caspase-3 substrate
was measured at 405 nm by a Microplate Absorbance Spectrophotometer
(Bio-Rad, Hercules, CA, USA). The detailed analysis procedure is
described in the manufacturer’s protocol.
Western blot analysis for assessment of
B-cell lymphoma 2 (Bcl-2) protein
Total cell lysates were separated using SDS-PAGE and
the proteins were transferred onto polyvinylidene fluoride
membranes (Invitrogen). The gels (Invitrogen)were blocked with 5%
non-fat dry milk in Tris-buffered saline (TBS) for 1 h at room
temperature and subsequently incubated overnight at 4°C with
primary antibodies (rabbit anti-mouse monoclonal anti-Bcl-2
antibody, Abcam, Cambridge, UK; anti-β-actin, Cell Signaling
Technology, Inc., Danvers, MA, USA) at the appropriate dilution
recommended in the product datasheet. Following three washes for
10–15 min each with TBS [0.1% (v/v)] in Tween 20 (TBST; Xiangya
Medical College), the gels were incubated with horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G (Cell
Signaling Technology, Inc.) in blocking buffer for 1 h at room
temperature. Following three washes with TBST, the gels were
developed with a chemiluminescence reagent (Cell Signaling
Technology, Inc.) and exposed to X-rays. The expression of Bcl-2
was semi-quantitatively evaluated by Gel-Pro Analyzer software,
version 4.5 (Media Cybernetics, Rockville, MD, USA) and compared
with the expression of β-actin.
Cholesterol efflux assays
Experiments were performed as described by Marcil
et al (15) with minor
modifications. Cells were planted at a density of 5×105
cells/ml in 24-well culture dishes. Following starvation for 24 h
in DMEM (high glucose) with 0.2% bovine serum albumin (BSA;
Invitrogen), cells were incubated in medium containing 0.5 μCi/well
[3H]-labeled cholesterol and 50 μg/ml ox-LDL for 48 h.
The cells were then incubated in the absence or presence of the
peptide Ac-hE-18A-NH2 at various concentrations (1, 10
and 50 μg/ml). The radioactivity in the supernatants and total cell
extracts was measured by scintillation counting. The efflux was
quantified as the percentage of total radioactive counts removed
from the cells during the efflux period.
Cellular cholesterol/cholesteryl ester
quantitation analysis
Cellular cholesterol/cholesteryl ester content was
measured using a commercially available quantitation kit
(Cholesterol/Cholesteryl Ester Quantitation Colorimetric Kit II;
BioVision, Inc., Mountain View, CA, USA), following the
manufacturer’s instructions.
Statistical analysis
All results are presented as the mean ± standard
deviation. Statistical analysis was performed using SPSS 13.0
software (SPSS, Inc., Chicago, IL, USA). Differences between the
groups were assessed by one-way analysis of variance. A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Ac-hE-18A-NH2 does not affect
the viability of RAW264.7 cells
The potential cytotoxic effect of the peptide
Ac-hE-18A-NH2 on the viability of RAW264.7 cells was
evaluated. There was no cytotoxic activity induced by the peptide
at the concentrations examined (1, 10, 50 and 100 μg/ml) within 24
h (data not shown).
Ox-LDL-induced apoptosis of RAW264.7
cells and effect of Ac-hE-18A-NH2 on ox-LDL-induced cell
apoptosis
Ox-LDL induced apoptosis in RAW264.7 cells in a
time- and concentration-dependent manner in the present study
(Table I). In the absence of
ox-LDL, a rate of apoptosis of merely 3.39±0.35% was detected
following 48 h of incubation. Compared with the control group, a
6.5-fold increase in the rate of apoptosis in the 50 μg/ml ox-LDL
group was observed, whereas a 12.2-fold increase was observed in
the 100-μg/ml ox-LDL group following 48 h of incubation (Table I). Significant levels of apoptosis
were obtained with ox-LDL treatment (50 μg/ml) for 24 h (24.4%) and
48 h (40.4%) (Table I).
 | Table IOx-LDL-induced apoptosis of RAW264.7
cells. |
Table I
Ox-LDL-induced apoptosis of RAW264.7
cells.
| Group | % apoptotic
cells |
|---|
| Control-24 h | 2.57±0.25 |
| Control-48 h | 3.39±0.35 |
| Ox-LDL 50 μg/ml-12
h | 13.69±0.66 |
| Ox-LDL 50 μg/ml-24
h | 24.40±2.57 |
| Ox-LDL 50 μg/ml-48
h | 40.36±3.37 |
| Ox-LDL 100 μg/ml-48
h | 70.74±4.35 |
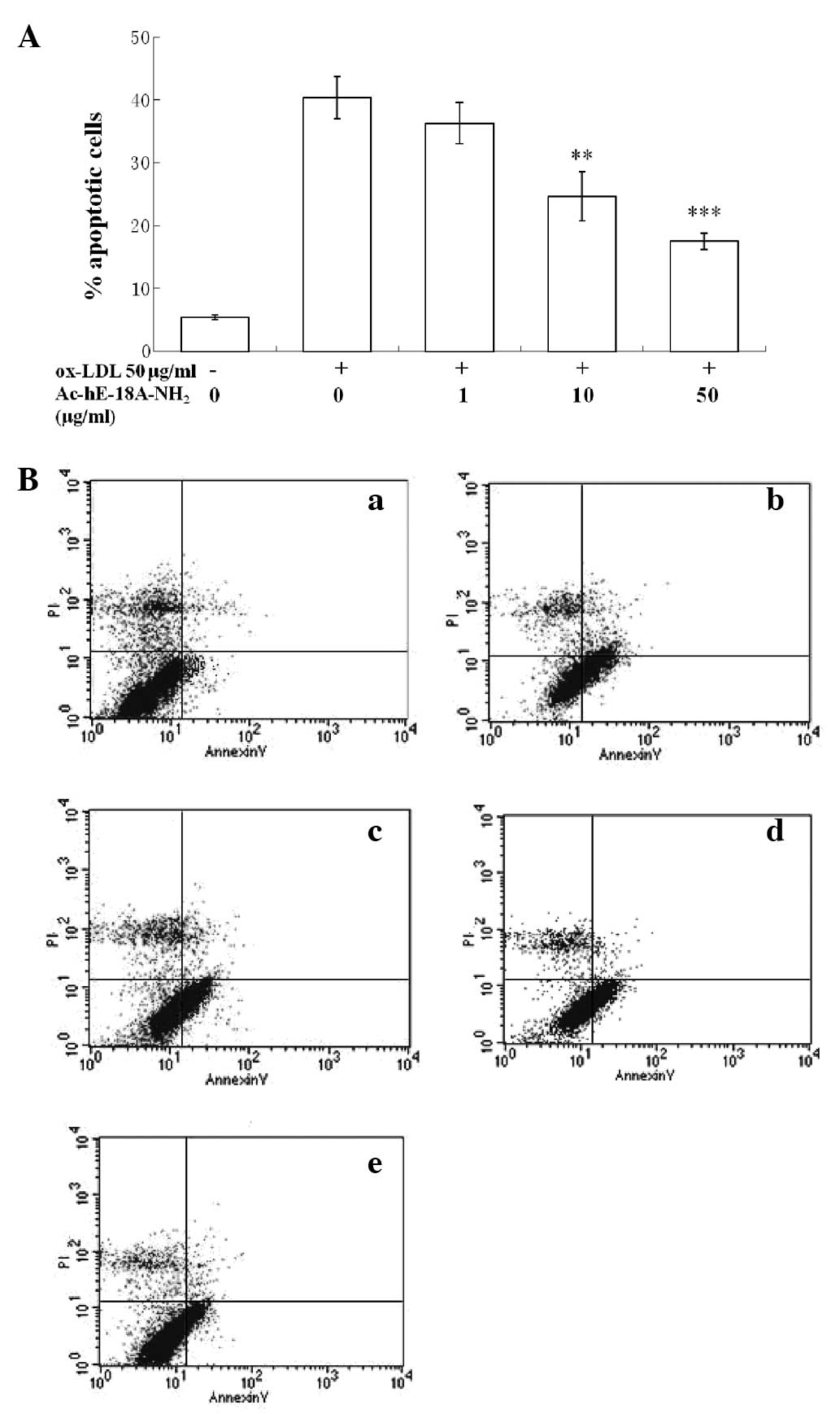
Incubation of macrophages with
Ac-hE-18A-NH2 for 24 h significantly reduced the toxic
effect of ox-LDL, and the protective effect was dose-dependent at
≤50 μg/ml (Fig. 1).
Effect of Ac-hE-18A-NH2 on
caspase-3 activity and Bcl-2 protein expression
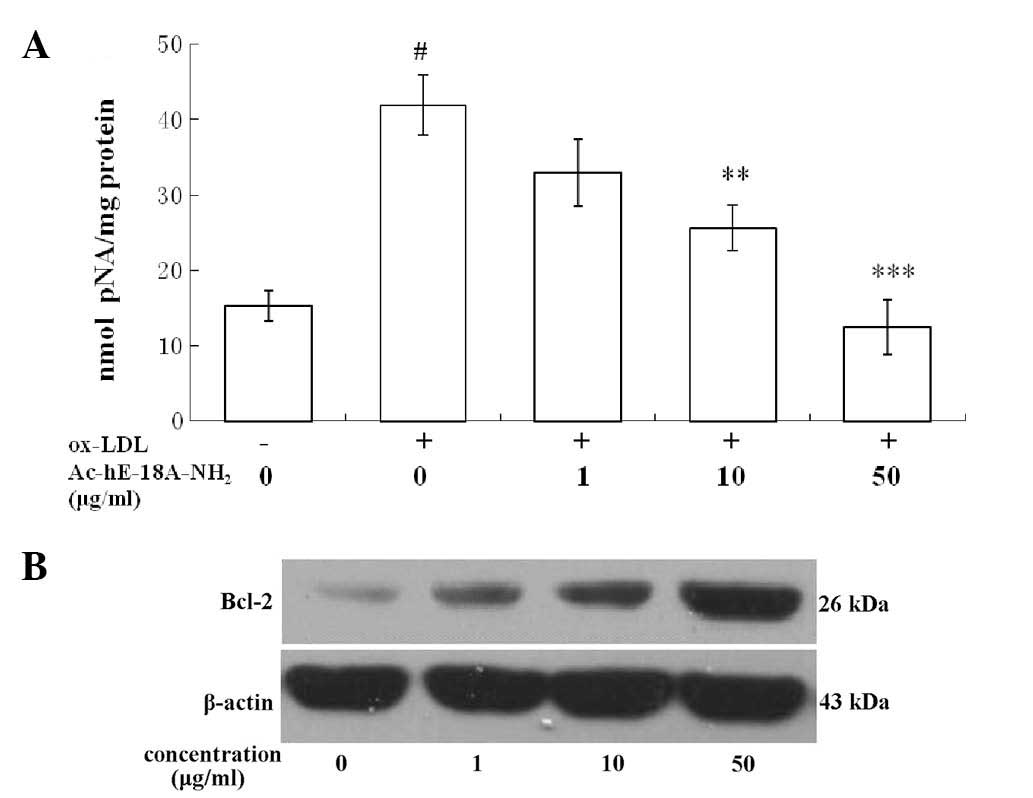
The activation of caspase-3 was analyzed by
measuring the levels of pNA cleaved from the substrate
N-Ac-DEVD-pNA. The activity assay showed that, following exposure
to 50 μg/ml ox-LDL for 48 h, Ac-DEVD-pNA cleavage was significantly
reduced in the macrophages treated with the peptide
Ac-hE-18A-NH2 compared with that in the cells that were
not treated with the peptide. Furthermore, Ac-hE-18A-NH2
reduced ox-LDL-induced caspase-3 activity in a dose-dependent
manner (Fig. 2A).
Bcl-2 protein expression was confirmed by western
blot analysis. The results indicated that exposure of RAW264.7
cells to ox-LDL reduced Bcl-2 protein expression, while treatment
with Ac-hE-18A-NH2 increased Bcl-2 protein expression
levels in a concentration-dependent manner (Fig. 2B).
Effect of Ac-hE-18A-NH2 on
cholesterol efflux and cholesterol levels in RAW264.7 cells
Exposure of RAW264.7 cells to ox-LDL for 48 h
increased total cellular cholesterol levels from 201.2±22.2 mg/g
protein to 590.6±23.3 mg/g protein without significantly affecting
the rate of cholesterol efflux (Table
II). Co-treatment with Ac-hE-18A-NH2 and ox-LDL
increased the rate of cholesterol efflux (Fig. 3A), and decreased the cellular total
cholesterol level (Table II). The
two alterations were concentration-dependent. In addition, apo A-I
was selected as a positive control in the efflux experiment in
order to evaluate the efficiency of the peptide. Of note,
Ac-hE-18A-NH2 (50 mg/ml) was more efficient than apo A-I
(10 mg/ml) in inducing cholesterol efflux (Fig. 3B).
| Table IIEffect of Ac-hE-18A-NH2 on
total cholesterol level in RAW264.7 cells. |
Table II
Effect of Ac-hE-18A-NH2 on
total cholesterol level in RAW264.7 cells.
| Group | Total cholesterol
(mg/g protein) |
|---|
| Control | 201.2±22.2 |
| Ox-LDL | 590.6±23.3a |
|
Ox-LDL+Ac-hE-18A-NH2 1
μg/ml | 490.5±12.8b |
|
Ox-LDL+Ac-hE-18A-NH2 10
μg/ml | 415.0±17.8b |
|
Ox-LDL+Ac-hE-18A-NH2 50
μg/ml | 299.2±16.4c |
Effect of brefeldin A (BFA) and
β-cyclodextrin (β-CD) on apoptosis and cholesterol efflux in
RAW264.7 cells
To further investigate the role of cholesterol
efflux on apoptosis induced by ox-LDL, β-CD and BFA were utilized.
β-CD has been shown to be capable of stimulating efficient
cholesterol efflux from cultured human fibroblasts (16). However, BFA, an antibiotic, has
been shown to inhibit high-density lipoprotein (HDL)-mediated
cholesterol efflux from cholesterol-enriched cells (17).
Flow cytometric analysis demonstrated that BFA alone
did not exert any effect on cellular apoptosis (data not shown).
However, both the inhibition of ox-LDL-induced apoptosis and the
facilitation of cholesterol efflux by Ac-hE-18A-NH2 (50
μg/ml) were significantly attenuated by BFA. Addition of β-CD
decreased the ox-LDL-induced rate of apoptosis from 17.53 to 10.89%
(Fig. 4B) and increased the rate
of cholesterol efflux from 25.56 to 43.03% (Fig. 4A).
Discussion
Ox-LDL has been shown to be taken up by macrophages
in a rapid and uncontrolled manner, leading to the formation of
cholesterol-loaded foam cells, the major cellular component of
fatty streaks (18). However,
ox-LDL may also modulate atherogenesis by inducing apoptosis in a
variety of tissues and cell types, including human coronary artery
endothelial cells (19), vascular
smooth muscle cells (20) and
monocyte-macrophages (21). Among
these, the apoptosis of macrophages has been proposed to be crucial
in the evolution of atherosclerotic plaques, and apoptotic
macrophages are often concentrated in areas of plaque rupture
(2). Furthermore, increasing
evidence suggests that ox-LDL and oxysterols are able to induce the
activation of the executioner caspase-3 (22) via the mitochondrial apoptotic
pathway. In addition, ox-LDL is able to promote the overexpression
of the Bcl-2-associated X-protein (Bax) (19) and reduce the expression of Bcl-2
(23), thereby promoting
susceptibility to apoptosis. The present study demonstrated that
exposure of macrophages to ox-LDL increases the apoptosis rate of
cells in a time- and dose-dependent manner. It was also observed
that ox-LDL downregulated Bcl-2 protein expression and promoted
caspase-3 activity.
Although the exact mechanism of ox-LDL-induced
apoptosis in macrophages remains to be elucidated, it has been
suggested that treatment with ox-LDL induces accumulation of large
amounts of cholesterol in macrophages and leads to failure of lipid
homeostasis (24). Furthermore,
cellular accumulation of excess cholesterol may serve as a trigger
of apoptosis and promote ongoing inflammation, calcification,
thrombosis and plaque rupture, which are the major sequelae of
advanced atherosclerosis. Plasma HDL levels are inversely
correlated with the risk of atherosclerotic cardiovascular disease
(25). One of the most important
atheroprotective roles of HDL is reverse cholesterol transport, in
which excess cholesterol in macrophage foam cells undergoes efflux
followed by being transported to the liver for excretion in the
bile. Terasaka et al (26)
demonstrated that HDL protects macrophages from FC- or
ox-LDL-induced apoptosis by promoting efflux of cholesterol via the
ATP binding cassette sub-family G member 1 (ABCG1) transporter
(26). In addition, Jiang et
al (27) reported that
HDL3 antagonizes ox-LDL-induced apoptosis in RAW264.7
cells through reducing the accumulation of toxic cholesterol
(28).
Recently, HDL/apo-based mimetic peptides have been
increasingly considered to be potential effective treatments for
atherosclerosis. Their atheroprotective activity is also attributed
to their unique ability to facilitate the reverse cholesterol
transport process. A previous study demonstrated that the
dual-domain peptide Ac-hE-18A-NH2 not only reduces
plasma cholesterol in dyslipidemic animal models but also exerts
anti-inflammatory/antioxidant effects through its ability to
scavenge ‘seeding molecules’ (28). The present study revealed that
Ac-hE-18A-NH2 inhibits apoptosis induced by ox-LDL in a
dose-dependent manner. Concomitantly, Ac-hE-18A-NH2
decreased caspase-3 activity and increased Bcl-2 expression in a
similarly concentration-dependent manner. An important question
that arose from this was whether the protection of macrophages from
ox-LDL-induced apoptosis by Ac-hE-18A-NH2 proceeds via
promoting the intracellular cholesterol efflux in a similar manner
to that of HDL. To elucidate this, cholesterol efflux and
intracellular cholesterol were assessed in macrophages treated with
ox-LDL and Ac-hE-18A-NH2. The results indicated that
Ac-hE-18A-NH2 dose-dependently increased the rate of
cholesterol efflux, and significantly decreased intracellulular
cholesterol levels in ox-LDL-treated RAW264.7 cells. However, it
was not elucidated whether the protective effect of this peptide on
ox-LDL-induced apoptosis was mediated via the promotion of
cholesterol efflux. To investigate this possibility, β-CD and BFA
were administered to stimulate or inhibit the cholesterol efflux
from macrophages. β-CD is a cyclic oligomer of glucose that has the
ability to sequester cholesterol in its hydrophobic core. A series
of studies have demonstrated that β-CD is particularly efficient in
stimulating the removal of cholesterol from a variety of cells in
culture (28,29). The exposure of cells to high
concentrations of β-CD (10–100 mM) results in rates of cholesterol
efflux far in excess of those achieved with physiological
cholesterol acceptors such as HDL. The selection of BFA as an
inhibitor was based on the fact that it strongly suppresses the
HDL/apo mediated cholesterol efflux from cholesterol-enriched cells
and it has been shown to affect intracellular trafficking of ABCA1
(30). Consistent with previous
studies, the present study has demonstrated that treatment of cells
with cholesterol acceptor β-CD promoted cholesterol efflux and
reduced the rate of ox-LDL-induced apoptosis. By contrast,
combination with BFA decreased the rate of cholesterol efflux
mediated by Ac-hE-18A-NH2 and increased the level of
total cholesterol, accompanied by an increase in ox-LDL-induced
apoptosis. This revealed the close correlation between apoptosis
and cholesterol accumulation induced by ox-LDL, and provided a
mechanism for the protection of macrophages by
Ac-hE-18A-NH2 from ox-LDL-induced apoptosis. Thus, the
results of this study support the hypothesis that the dual-domain
synthetic peptide Ac-hE-18A-NH2 may have therapeutic
potential for reducing common pathological features of
atherosclerosis. In addition, the ongoing investigation of the
minimized domain structure in apolipoproteins remains an
ever-important asset to the understanding of the functions of
apolipoproteins in atherogenesis and may yield new therapies for
ameliorating cardiovascular diseases.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 30770857) and the Key
Clinical Program of the Ministry of Health.
References
|
1
|
Seimon T and Tabas I: Mechanisms and
consequences of macrophage apoptosis in atherosclerosis. J Lipid
Res. 50(Suppl): S382–S387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tabas I: Consequences and therapeutic
implications of macrophage apoptosis in atherosclerosis: the
importance of lesion stage and phagocytic efficiency. Arterioscler
Thromb Vasc Biol. 25:2255–2264. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tabas I: Consequences of cellular
cholesterol accumulation: basic concepts and physiological
implications. J Clin Invest. 110:905–911. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kellner-Weibel G, Jerome WG, Small DM, et
al: Effects of intracellular free cholesterol accumulation on
macrophage viability: a model for foam cell death. Arterioscler
Thromb Vasc Biol. 18:423–431. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yao PM and Tabas I: Free cholesterol
loading of macrophages is associated with widespread mitochondrial
dysfunction and activation of the mitochondrial apoptosis pathway.
J Biol Chem. 276:42468–42476. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yao PM and Tabas I: Free cholesterol
loading of macrophages induces apoptosis involving the fas pathway.
J Biol Chem. 275:23807–23813. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anantharamaiah GM: Synthetic peptide
analogs of apolipoproteins. Methods Enzymol. 128:627–647. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Datta G, Chaddha M, Garber DW, et al: The
receptor binding domain of apolipoprotein E, linked to a model
class A amphipathic helix, enhances internalization and degradation
of LDL in fibroblasts. Biochemistry. 39:213–220. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Datta G, Garber DW, Chung BH, et al:
Cationic domain 141–150 of apoE covalently linked to a class A
amphipathic helix enhances atherogenic lipoprotein metabolism in
vitro and in vivo. J Lipid Res. 42:959–966. 2001.
|
|
10
|
Garber DW, Handattu S, Aslan I, Datta G,
Chaddha M and Anantharamaiah GM: Effect of an arginine-rich
amphipathic helical peptide on plasma cholesterol in dyslipidemic
mice. Atherosclerosis. 168:229–237. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gupta H, White CR, Handattu S, et al:
Apolipoprotein E mimetic Peptide dramatically lowers plasma
cholesterol and restores endothelial function in watanabe heritable
hyperlipidemic rabbits. Circulation. 111:3112–3118. 2005.
View Article : Google Scholar
|
|
12
|
Sharifov OF, Nayyar G, Garber DW, et al:
Apolipoprotein E mimetics and cholesterol-lowering properties. Am J
Cardiovasc Drugs. 11:371–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Navab M, Hama SY, Anantharamaiah GM, et
al: Normal high density lipoprotein inhibits three steps in the
formation of mildly oxidized low density lipoprotein: steps 2 and
3. J Lipid Res. 41:1495–1508. 2000.
|
|
14
|
Datta G, Epand RF, Epand RM, et al:
Aromatic residue position on the nonpolar face of class A
amphipathic helical peptides determines biological activity. J Biol
Chem. 279:26509–26517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Marcil M, Bissonnette R, Vincent J,
Krimbou L and Genest J: Cellular phospholipid and cholesterol
efflux in high-density lipoprotein deficiency. Circulation.
107:1366–1371. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yancey PG, Rodrigueza WV, Kilsdonk EP,
Stoudt GW, Johnson WJ, Phillips MC and Rothblat GH: Cellular
cholesterol efflux mediated by cyclodextrins. Demonstration of
kinetic pools and mechanism of efflux. J Biol Chem.
271:16026–16234. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mendez AJ: Monensin and brefeldin A
inhibit high density lipoprotein-mediated cholesterol efflux from
cholesterol-enriched cells. J Biol Chem. 270:5891–5900.
1995.PubMed/NCBI
|
|
18
|
Hung YC, Hong MY and Huang GS: Cholesterol
loading augments oxidative stress in macrophages. FEBS Letters.
580:849–861. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li D and Mehta JL: Upregulation of
endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL and
implications in apoptosis of human coronary artery endothelial
cells: evidence from use of antisense LOX-1 mRNA and chemical
inhibitors. Arterioscler Thromb Vasc Biol. 20:1116–1122. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kataoka H, Kume N, Miyamoto S, et al:
Oxidized LDL modulates Bax/Bcl-2 through the lectinlike Ox-LDL
receptor-1 in vascular smooth muscle cells. Arterioscler Thromb
Vasc Biol. 21:955–960. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hardwick SJ, Hegyi L, Clare K, et al:
Apoptosis in human monocyte-macrophages exposed to oxidized low
density lipoprotein. J Pathol. 179:294–302. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wintergerst ES, Jelk J, Rahner C and Asmis
R: Apoptosis induced by oxidized low density lipoprotein in human
monocyte-derived macrophages involves CD36 and activation of
caspase-3. Eur J Biochem. 267:6050–6059. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Napoli C, Quehenberger O, De Nigris F,
Abete P, Glass CK and Palinski W: Mildly oxidized low density
lipoprotein activates multiple apoptotic signaling pathways in
human coronary cells. FASEB J. 14:1996–2007. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yancey PG and Jerome WG: Lysosomal
sequestration of free and esterified cholesterol from oxidized low
density lipoprotein in macrophages of different species. J Lipid
Res. 39:1349–1361. 1998.PubMed/NCBI
|
|
25
|
Sanossian N, Saver JL, Navab M and
Ovbiagele B: High-density lipoprotein cholesterol: an emerging
target for stroke treatment. Stroke. 38:1104–1109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Terasaka N, Wang N, Yvan-Charvet L and
Tall AR: High-density lipoprotein protects macrophages from
oxidized low-density lipoprotein-induced apoptosis by promoting
efflux of 7-ketocholesterol via ABCG1. Proc Natl Acad Sci USA.
104:15093–15098. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang P, Yan PK, Chen JX, et al: High
density lipoprotein 3 inhibits oxidized low density
lipoprotein-induced apoptosis via promoting cholesterol efflux in
RAW264.7 cells. Acta Pharmacol Sin. 27:151–157. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Christian AE, Haynes MP, Phillips MC and
Rothblat GH: Use of cyclodextrins for manipulating cellular
cholesterol content. J Lipid Res. 38:2264–2272. 1997.PubMed/NCBI
|
|
29
|
Tsujikawa H, Song Y, Watanabe M, et al:
Cholesterol depletion modulates basal L-type Ca2+
current and abolishes its -adrenergic enhancement in ventricular
myocytes. Am J Physiol Heart Circ Physiol. 294:H285–H292. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Verghese PB, Arrese EL, Howard AD and
Soulages JL: Brefeldin A inhibits cholesterol efflux without
affecting the rate of cellular uptake and re-secretion of
apolipoprotein A-I in adipocytes. Arch Biochem Biophys.
478:161–166. 2008. View Article : Google Scholar : PubMed/NCBI
|