Introduction
Gap junctions connect the interior of cells and are
important in the maintenance of tissue homeostasis. Certain
deleterious metabolites involved in cerebral ischemia and apoptotic
associated molecules, including Ca2+, IP3,
ATP and cAMP, may be small enough to pass through gap junctions and
modulate cell death of the neighboring cells, enhancing the
spreading of injury (1,2). Alternatively, the network of gap
junctions could prevent the death of injured cells by the buffering
of toxic metabolites to healthy surrounding cells (3). Thus, gap junctions could have
damaging and protective effects (4).
There is increasing evidence that suppressing gap
junctions is able to reduce the spread of damage following ischemia
(5). For example, the gap junction
blocker octanol restricted the flow of undesirable neurotoxins and
significantly decreased the spread of cell death (6). Similarly, connexin 43 (the
predominant connexin in gap junctions) heterozygous mice
demonstrated reduced shrinkage and metabolite abnormality in the
ipsilateral hippocampus following middle cerebral artery occlusion
(MCAO) (7). Lin et al
demonstrated in vitro that the resistance of C6 cells to
calcium overload and oxidative stress was compromised when they
formed gap junctions with more vulnerable cells (8). Despite the detrimental role of gap
junctions, it is noteworthy that gap junctions may mediate survival
or be protective for adjacent cells. In mixed astrocyte neuron
cultures, the inhibition of astrocyte coupling with gap junction
blockers increased neuronal vulnerability to oxidative stress or
glutamic acid toxicity (9,10). Consistently, connexin 43
overexpression substantially suppressed zinc (released during
ischemia) toxicity whereas its knockdown caused a significant
enhancement of the toxicity (11).
Certain investigators proposed that the opposite
role of gap junctions may be associated with the extent of neuronal
death (12), which was highly
correlated with the duration of ischemia (13,14).
Therefore, the present study examined whether the opposite role of
gap junctions was associated with occlusion time. In order to
examine this issue, a gap junction blocker octanol was used, which
has been administered to investigate the role of gap junctions in
numerous studies (6,15). The effects of octanol in MCAO was
evaluated using neurological deficits, infarct volume and
transferase dUTP nick-end labeling (TUNEL) staining. To further
investigate the mechanism of the opposite roles of octanol, the
expression of Bcl-2 and Bax was studied, which play opposite roles
in apoptosis (16). The present
study provides evidence that may be used to further elucidate the
opposite role of gap junctions in ischemia.
Materials and methods
Animal model of cerebral ischemia
Male adult Sprague-Dawley rats weighing 250–280 g
were maintained in a temperature- and light-controlled environment
with a 12 h light/dark cycle. All experiments were performed in
accordance with the NIH guidelines and approval by the Animal Care
and Use Committee of the Guangzhou University of Chinese Medicine
(Guangzhou, Guangdong, China) and all efforts were made to minimize
animal suffering.
A focal cerebral ischemic rat model was induced by
MCAO as previously described (17). In brief, the rats were anesthetized
with 350 mg/kg of chloral hydrate via intraperitoneal injection.
The right carotid bifurcation was exposed and the external carotid
artery was coagulated distal to the bifurcation. A 4–0 nylon
monofilament suture with a rounded tip was introduced into the
internal carotid artery through the stump of the external carotid
artery and gently advanced for ~20 mm. Following 30 min/2 h of
occlusion, the filament was gently withdrawn and the incision was
closed (18). During the whole
process, the rat rectal temperature was maintained at 37°C by
placing the animals on a heating bed. In the sham-operated rats,
the common carotid was exposed without ligation.
Drug treatment
Octanol purchased from Sigma (St. Louis, MO, USA)
was dissolved in dimethyl sulfoxide (DMSO; 0.005% v/v) and a final
concentration of 5 mmol/kg was administered intraperitoneally 30
min prior to ischemia (6). The
vehicle-treated rats were administered equal volumes of DMSO.
Measurement of neurological deficits
An independent observer performed the assessment of
the neurological deficit score: 0, no neurological deficit; 1,
failure to fully extend left forepaw; 2, circling to the left; 3,
falling to the left; 4, loss of spontaneous walking with a
depressed level of consciousness and 5, dead (17).
Measurement of infarct volume
Following reperfusion for 24 h, rats were overdosed
with sodium pentobarbital (100 mg/kg). The brains were quickly
removed and cut into coronal slices of 2 mm in thickness. The
sections were immersed in a 2% solution of
2,3,5-triphenyltetrazolium chloride (TTC; Mym Biological
Technology, Andhra Pradesh, India) for 20 min at 37°C and then
fixed with 4% paraformaldehyde. The infarct area was measured by
each slice using a computerized image analysis system
(Image-Pro-Plus; Media Cybernetics, Silver Spring, MD, USA).
Infarct areas of all sections were added to derive the total
infarct area, which was multiplied by the thickness. The infarct
volume was expressed as a percentage of the ipsilateral hemispheric
volume (%).
Tissue preparation and TUNEL assay
Rats were deeply anesthetized with sodium
pentobarbital and perfused through the ascending aorta with cold
saline, followed by 4% paraformaldehyde in 0.1 M of
phosphate-buffered saline (PBS; pH 7.2–7.4, 4°C). The brains were
cytoprotected by 15 and 30% sucrose sequentially. Coronal sections
were cut on a freezing microtome at a thickness of 15 μm starting
at +1.60 mm to −4.80 mm from the bregma and then were mounted onto
polylysine-coated slides. Intervening sections were air dried, then
stored at −80°C until future use. TUNEL staining was processed
using an in situ cell death detection kit (Roche Molecular
Biochemicals, Mannheim, Germany) according to the manufacturer’s
instructions. Images were captured using an Olympus microscope
(Olympus, Tokyo, Japan) and the number of TUNEL-positive neurons
within the CA1 subfield was counted.
Immunohistochemistry
Brain sections (15 μm thickness) were mounted with
2% goat serum in 0.1 M of Tris buffered saline (TBS)/0.3% Triton
X-100 for 1 h at room temperature and incubated overnight at 4°C
with the primary antibody for Bcl-2/Bax (1:100; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA). Following washing in
TBS, sections were incubated with biotinylated goat anti-rabbit IgG
and further processed with the ABC method (Vector, San Diego, CA,
USA). The stained sections were captured with a CCD spot camera.
Non-specific staining was determined by omitting the primary
antibodies.
To quantify Bcl-2 and Bax immunoreactivity (IR), the
measurement was performed with a computerized image analysis system
(Image-Pro-Plus, Media Cybernetics) according to the previously
described method (19). Briefly, a
density threshold was set above the background level firstly to
identify a positively stained structure and the area occupied by
these structures was measured as positive area. An average
percentage of Bcl-2-IR or Bax-IR area to the total outlined area
was obtained. Five to six animals were included in each group for
the quantification of immunohistochemistry.
Quantification and statistical
analysis
Observers blinded to the experimental conditions,
evaluated the outcome measures. All data are presented as the mean
± SEM. The Student’s t-test was used to compare the difference
between the vehicle group and the octanol group. All analysis was
performed using the Statistical Package for Social Sciences (SPSS,
version 13.0 for Windows). P<0.05 was considered to indicate a
statistically significant difference.
Results
Neurological deficits
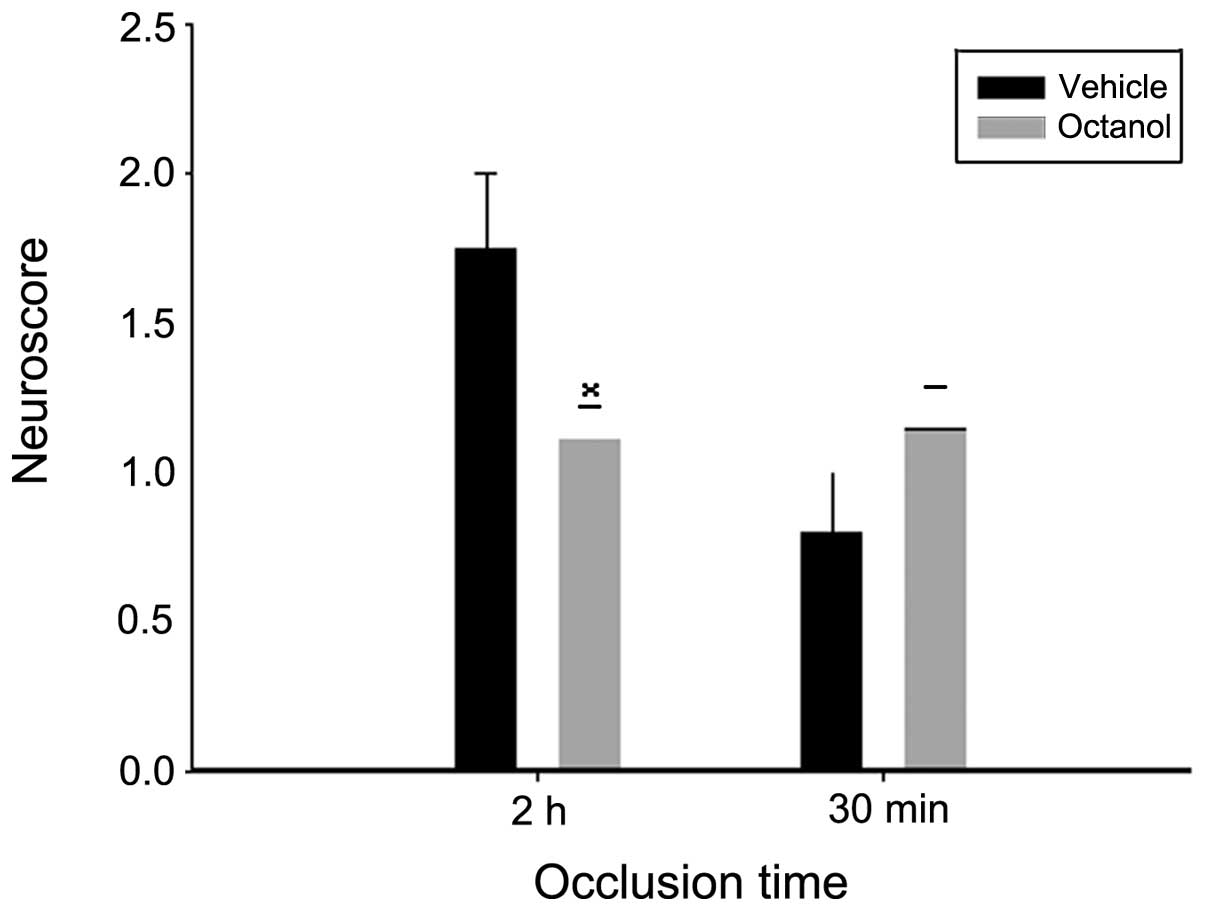
The effect of octanol was evaluated by the Zea Longa
test. As shown in Fig. 1, octanol
significantly lowered the neurological deficits compared with the
vehicle group following 2 h of ischemia and 24 h of reperfusion
(P=0.028). As for the 30 min ischemia group, although the mean of
the neurological deficit scores was higher in the octanol group,
the difference between the two groups was not statistically
significant (P=0.181).
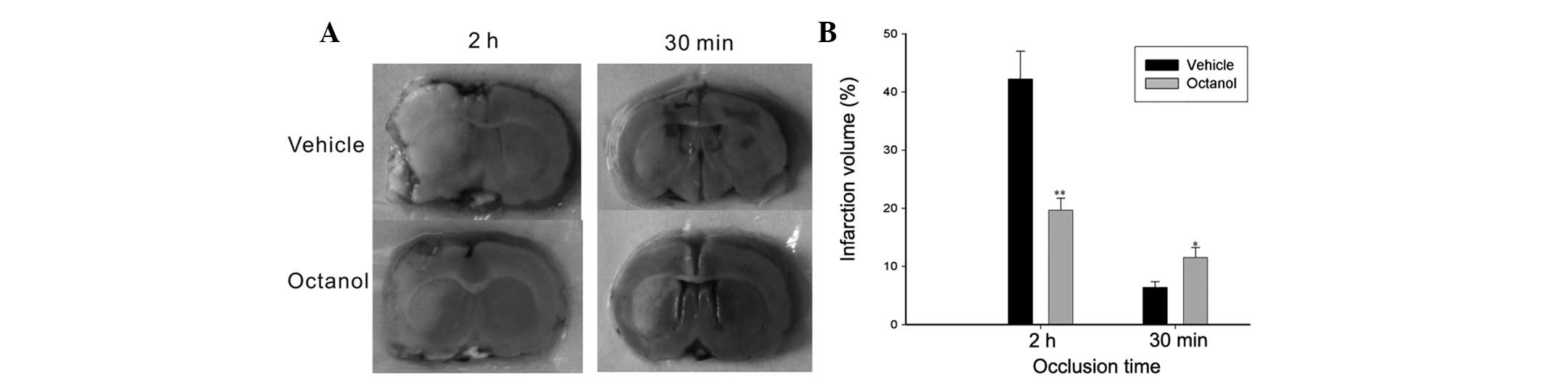
Infarct volume evaluation
Representative images of TTC staining were shown in
Fig. 2. The infarct volume
following 2 h of occlusion was reduced by octanol pretreatment
compared with the vehicle group (P=0.001). Conversely, a larger
stroke volume was observed in the octanol group following 30 min of
occlusion compared with that in the vehicle group (P=0.025).
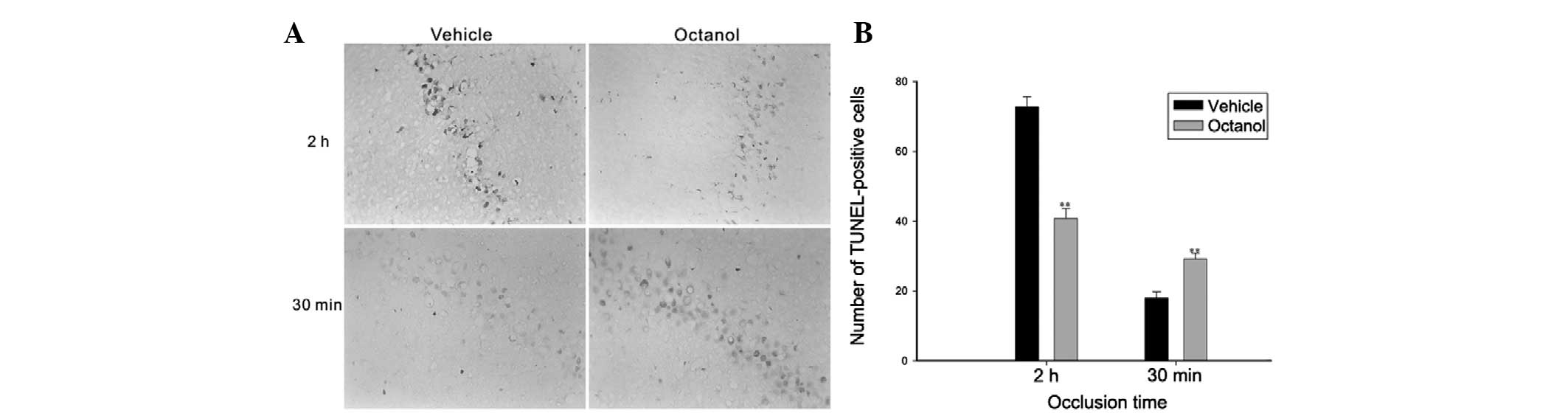
TUNEL assay
A moderate number of markedly TUNEL-labeled CA1
pyramidal neurons were identified in the CA1 region in the vehicle
group following ischemia for 2 h and reperfusion for 24 h. However,
the number of TUNEL-positive neurons in the CA1 region of rats
pretreated with octanol was markedly decreased (t=7.812,
P<0.001; Fig. 3). Notably, in
the 30 min ischemia group, octanol significantly increased the
number of TUNEL-positive cells compared with the vehicle group
(t=4.595, P=0.002; Fig. 3).
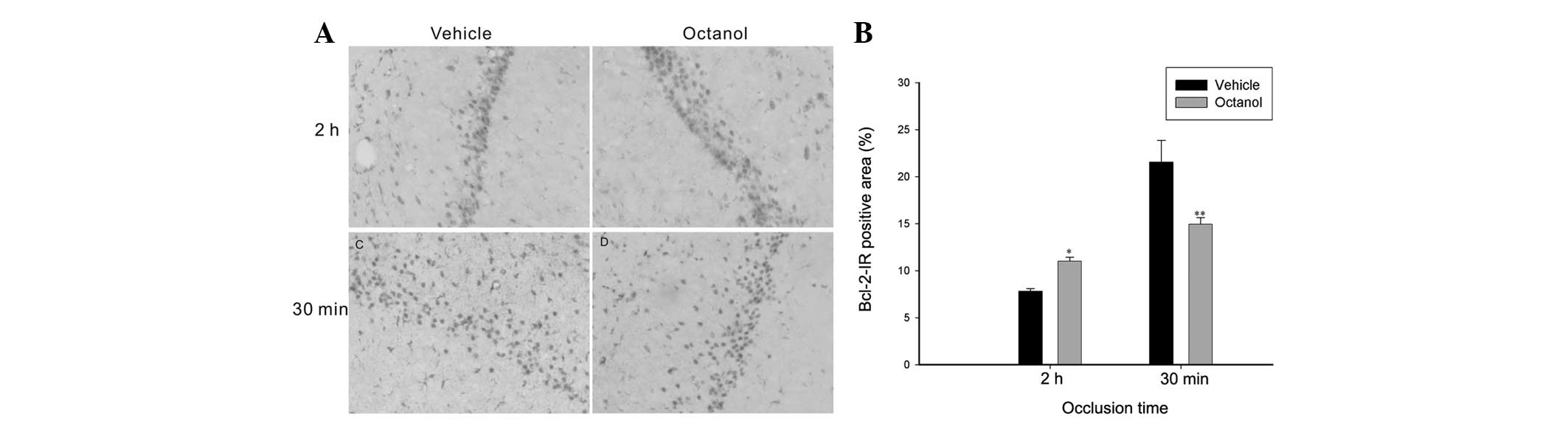
Expression of Bcl-2 and Bax
Apoptosis contributes to delayed neuronal death
causing expansion of the stroke lesion, therefore we investigated
the expression of anti-apoptotic Bcl-2 and pro-apoptotic Bax
expression. In the 2 h ischemia group, compared with the vehicle
group, octanol significantly increased the expression of Bcl-2
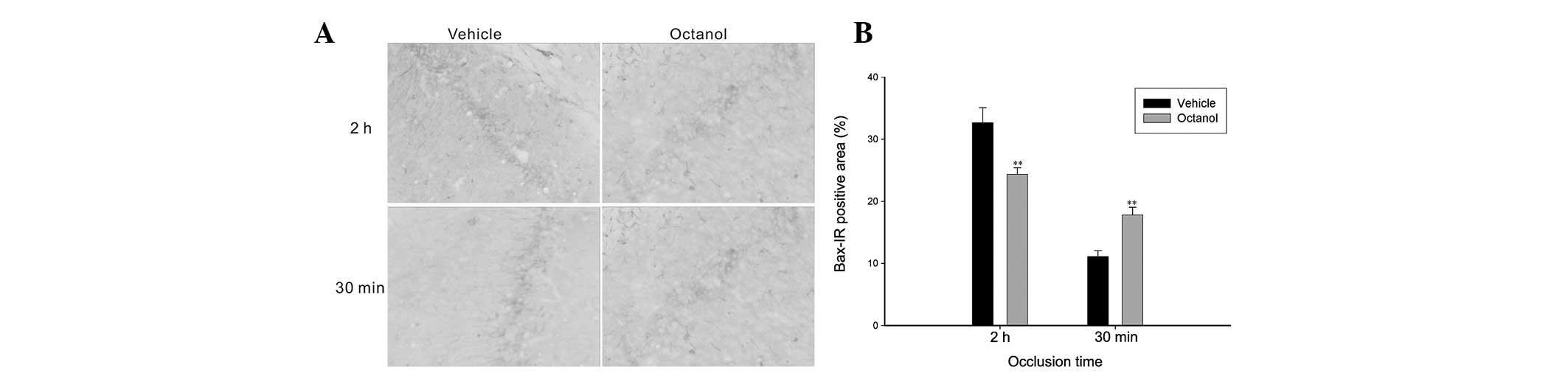
(t=6.151, P<0.001), however decreased the expression of Bax
(t=2.768, P=0.016; Figs. 4 and
5). However, in the 30 min
ischemia group, the rats treated with octanol demonstrated that,
accompanied by a decrease in Bcl-2 expression (t=2.907, P=0.012),
Bax expression was markedly increased (t=4.149, P=0.001; Figs. 4 and 5).
Discussion
The present study demonstrated and compared for the
first time, to the best of our knowledge, the effect of the gap
junction blocker octanol in MCAO for 2 h and 30 min, respectively.
Compared with the vehicle-treated rats, octanol attenuated the
ischemia injury induced by 2 h of occlusion demonstrated by the
neurological deficit, infarct volume and TUNEL staining, whereas it
aggravated the ischemia injury induced by 30 min of occlusion,
indicating that octanol was protective following 2 h of occlusion
but harmful following 30 min of occlusion. Octanol, an eight-carbon
aliphatic alcohol, is a relatively specific inhibitor of gap
junction permeability and is frequently used to study the role of
gap junctions. Although certain studies have demonstrated that
octanol can interfere with synaptic transmission (20), it is hard to interpret these two
opposite effects with gap junction-independent mechanisms. The
opposite role of octanol appears to be closely associated with the
function of gap junctions (21).
For example, gap junctions could be neuroprotective since adjacent
healthy cells may act as an effective spatial buffer against the
extracellular accumulation of neurotoxic substances (22,23).
By contrast, they could also be harmful when neighboring cells are
unable to aid in the clearance of the neurotoxic substances, which
could depolarize a large number of neurons/glia promoting the
release of glutamate and therefore causing aggravated injury
(24).
Gap junctions have been revealed to be protective in
the model of hypoxic preconditioning (subthreshold insults)
(25), MCAO above and below the
rhinal fissure (26–29) and MCAO for 30 min (30). By contrast, previous investigations
also demonstrated that gap junctions were harmful in the model of
four vessels occlusion (31),
clamping the common carotid arteries and lowering the mean arterial
blood pressure to 40 mmHg (6),
MCAO for 16 h (32) and
oxygen-glucose deprivation (OGD) for 6 to 10 days (33). As the intensity and/or the duration
of the ischemic episode increases, the predominant function of gap
junctions may be different. For example, when the buffering effect
is stronger than the harmful factors’ propagation in the condition
that injury is mild, gap junctions may be neuroprotective;
otherwise gap junctions may be harmful. In the present study, the
infarct volume following 30 min of occlusion was only 6.4%, thus
gap junctions were efficient in buffering harmful factors. By
contrast, 2 h of occlusion induced a much larger infarct volume,
therefore gap junctions could not buffer harmful factors
efficiently and more neurotoxic substances propagated to
neighbouring cells aggravating the injury instead.
Notably, while octanol significantly increased the
infarct volume and cell death following 30 min of occlusion, no
statistical differences in neurological deficits were identified
between the octanol group and the vehicle group. Similarly, Longa
et al demonstrated that the infarct area following 2 h of
temporary MCAO were 15.7% smaller than that following permanent
MCAO, however the neurological deficit was not significantly
reduced (17). In light of these
findings, the neurological score was not closely associated with
the infarct volume as occasionally the changes in infarct volume
may not be enough to create differences in the neurological
test.
To explore the possible mechanisms, we detected
Bcl-2 and Bax immunoreactivity, respectively, which are two
opposite apoptotic factors. Bcl-2 can help neuronal survival and
protect brain tissue from ischemic injury (34,35).
It acted upstream to prevent the activation of caspases, inhibited
free radical formation and regulated calcium sequestration
(36). However, Bax inhibition
reduced apoptotic neuronal injury in the hippocampal CA1 region and
behavioral deficits following global ischemia (37,38).
In the present study, we revealed that octanol induced Bcl-2
expression and attenuated Bax expression in MCAO for 2 h. By
contrast, it inhibited Bcl-2 expression and increased Bax staining
in MCAO for 30 min. These results demonstrated that the protective
role of octanol in ischemia for 2 h may be associated with its
ability to prevent apoptosis via facilitation of the anti-apoptotic
factor Bcl-2 or inhibition of the apoptotic biochemical factor Bax.
Conversly, the decreased Bcl-2 and increased Bax contributed to
apoptosis, thus resulting in a harmful role of octanol in ischemia
for 30 min. We are not completely certain that the opposite roles
of octanol on Bcl-2 and Bax is directly mediated by gap junction
activity. However, the consistency between ischemia injury and the
corresponding Bcl-2/Bax expression in ischemia for different
durations, to a certain extent, verifies that the present results
are mainly attributed to the effect of octanol on gap
junctions.
In summary, the present study investigated and
compared the effects of octanol in MCAO for the first time, to the
best of our knowledge. Our results demonstrated that the gap
junction blocker octanol played opposite roles in MCAO for
different durations, which was demonstrated by the neurological
outcome, infarct size and TUNEL staining. In addition, the function
of octanol was at least in part mediated through the
counter-regulation of Bcl-2 and Bax. These data provide a novel
perspective for the role of gap junctions in cerebral ischemia and
may be beneficial for the potential clinical treatment of various
ischemic conditions. However, more studies are required to examine
this, including using other blockers of gap junctions or connexin
43 deficient mice.
Acknowledgements
This study was supported by the National Nature
Science Foundation (no. 81202817), the Guangdong Province Medical
Research Foundation (no. B2011123), the Natural Science Foundation
of Guangdong Province (no. S2012040007519) and the Guangdong
College of outstanding youth innovation talent training project
(no. LYM11042; no. 2012LYM0045).
Abbreviations:
|
MCAO
|
middle cerebral artery occlusion
|
|
OGD
|
oxygen-glucose deprivation
|
|
TUNEL
|
transferase dUTP nick-end labeling
|
|
IR
|
immunoreactive
|
|
Bcl-2
|
B-cell leukemia-2
|
|
Bax
|
Bcl-2-associated X
|
References
|
1
|
Krysko DV, Leybaert L, Vandenabeele P and
D’Herde K: Gap junctions and the propagation of cell survival and
cell death signals. Apoptosis. 10:459–469. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Peixoto PM, Ryu SY, Pruzansky DP,
Kuriakose M, Gilmore A and Kinnally KW: Mitochondrial apoptosis is
amplified through gap junctions. Biochem Biophys Res Commun.
390:38–43. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sahores M and Mendoza-Naranjo A: Gap
junctions as therapeutic targets in brain injury following
hypoxia-ischemia. Recent Pat CNS Drug Discov. 3:209–315. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spray DC, Hanstein R, Lopez-Quintero SV,
Stout RF Jr, Suadicani SO and Thi MM: Gap junctions and Bystander
effects: Good Samaritans and executioners. Wiley Interdiscip Rev
Membr Transp Signal. 2:1–15. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Davidson JO, Green CR, Bennel L, Nicholson
LF, Danesh-Meyer H, O’Carroll SJ and Gunn AJ: A key role for
connexin hemichannels in spreading ischemic brain injury. Curr Drug
Targets. 14:36–46. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rami A, Volkmann T and Winckler J:
Effective reduction of neuronal death by inhibiting gap junctional
intercellular communication in a rodent model of global transient
cerebral ischemia. Exp Neurol. 170:297–304. 2001. View Article : Google Scholar
|
|
7
|
Xie M, Yi C, Luo X, et al: Glial gap
junctionsal communication involvement in hippocampal damage after
middle cerebral artery occlusion. Ann Neurol. 70:121–132. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin JH, Weigel H, Cotrina ML, et al:
Gap-junction-mediated propagation and amplification of cell injury.
Nat Neurosci. 1:494–500. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Blanc EM, Bruce-Keller AJ and Mattson MP:
Astrocytic gap junctional communication decreases neuronal
vulnerability to oxidative stress-induced disruption of
Ca2+ homeostasis and cell death. J Neurochem.
70:958–970. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ozog MA, Siushansian R and Naus CC:
Blocking gap junctional coupling increases glutamate-induced
neurotoxicity in neuron-astrocyte co-cultures. J Neuropathol Exp
Neurol. 61:132–141. 2002.PubMed/NCBI
|
|
11
|
Lee J, Yim YS, Ko SJ, Kim DG and Kim CH:
Gap junctions contribute to astrocytic resistance against zinc
toxicity. Brain Res Bull. 86:314–318. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chew SS, Johnson CS, Green CR and
Danesh-Meyer HV: Role of connexin43 in central nervous system
injury. Exp Neurol. 225:250–261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bendel O, Alkass K, Bueters T, von Euler M
and von Euler G: Reproducible loss of CA1 neurons following carotid
artery occlusion combined with halothane-induced hypotension. Brain
Res. 1033:135–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Colbourne F, Li H and Buchan AM:
Continuing postischemic neuronal death in CA1: influence of
ischemia duration and cytoprotective doses of NBQX and SNX-111 in
rats. Stroke. 30:662–668. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rawanduzy A, Hansen A, Hansen TW and
Nedergaard M: Effective reduction of infarct volume by gap junction
blockade in a rodent model of stroke. J Neurosurg. 87:916–920.
1997.
|
|
16
|
Kaido T, Kempski O, Heimann A, Heers C and
Bartsch D: Cluster analysis of mRNA expression levels identifies
multiple sequential patterns following focal cerebral ischemia.
Turk Neurosurg. 22:441–447. 2012.
|
|
17
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zivin JA: Factors determining the
therapeutic window for stroke. Neurology. 50:599–603. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
George A, Marziniak M, Schäfers M, Toyka
KV and Sommer C: Thalidomide treatment in chronic constrictive
neuropathy decreases endoneurial tumor necrosis factor-alpha,
increases interleukin-10 and has long-term effects on spinal cord
dorsal horn met-enkephalin. Pain. 88:267–275. 2000. View Article : Google Scholar
|
|
20
|
Narahashi T, Aistrup GL, Lindstrom JM,
Marszalec W, Nagata K, Wang F and Yeh JZ: Ion channel modulation as
the basis for general anesthesia. Toxicol Lett. 100:185–191. 1998.
View Article : Google Scholar
|
|
21
|
Contreras JE, Sánchez HA, Véliz LP,
Bukauskas FF, Bennett MV and Sáez JC: Role of connexin-based gap
junction channels and hemichannels in ischemia-induced cell death
in nervous tissue. Brain Res Brain Res Rev. 47:290–303. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Retamal MA, Schalper KA, Shoji KF,
Orellana JA, Bennett MV and Sáez JC: Possible involvement of
different connexin43 domains in plasma membrane permeabilization
induced by ischemia-reperfusion. J Membr Biol. 218:49–63. 2007.
View Article : Google Scholar
|
|
23
|
Rouach N, Avignone E, Même W, Koulakoff A,
Venance L, Blomstrand F and Giaume C: Gap junctions and connexin
expression in the normal and pathological central nervous system.
Biol Cell. 94:457–475. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Talhouk RS, Zeinieh MP, Mikati MA and
El-Sabban ME: Gap junctional intercellular communication in
hypoxia-ischemia-induced neuronal injury. Prog Neurobiol. 84:57–76.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin JH, Lou N, Kang N, et al: A central
role of connexin 43 in hypoxic preconditioning. J Neurosci.
28:681–695. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakase T, Fushiki S and Naus CC:
Astrocytic gap junctions composed of connexin 43 reduce apoptotic
neuronal damage in cerebral ischemia. Stroke. 34:1987–1993. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakase T, Fushiki S, Söhl G, Theis M,
Willecke K and Naus CC: Neuroprotective role of astrocytic gap
junctions in ischemic stroke. Cell Commun Adhes. 10:413–417. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakase T, Sähl G, Theis M, Willecke K and
Naus CC: Increased apoptosis and inflammation after focal brain
ischemia in mice lacking connexin43 in astrocytes. Am J Pathol.
164:2067–2075. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Siushansian R, Bechberger JF, Cechetto DF,
Hachinski VC and Naus CC: Connexin43 null mutation increases
infarct size after stroke. J Comp Neurol. 440:387–394. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang F, Hai J and Jing Y: Gap junctions
communication involved in brain protection following focal ischemia
and reperfusion in rats. Neural Regen Res. 9:677–682. 2009.(In
Chinese).
|
|
31
|
Perez Velazquez JL, Kokarovtseva L,
Sarbaziha R, Jeyapalan Z and Leshchenko Y: Role of gap junctionsal
coupling in astrocytic networks in the determination of global
ischaemia-induced oxidative stress and hippocampal damage. Eur J
Neurosci. 23:1–10. 2006.PubMed/NCBI
|
|
32
|
Saito R, Graf R, Hübel K, Fujita T, Rosner
G and Heiss WD: Reduction of infarct volume by halothane: effect on
cerebral blood flow or perifocal spreading depression-like
depolarizations. J Cereb Blood Flow Metab. 17:857–864.
1997.PubMed/NCBI
|
|
33
|
de Pina-Benabou MH, Szostak V, Kyrozis A,
et al: Blockade of gap junctions in vivo provides neuroprotection
after perinatal global ischemia. Stroke. 36:2232–2237.
2005.PubMed/NCBI
|
|
34
|
Martinou JC, Dubois-Dauphin M, Staple JK,
et al: Overexpression of BCL-2 in transgenic mice protects neurons
from naturally occurring cell death and experimental ischemia.
Neuron. 13:1017–1030. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen J, Simon RP, Nagayama T, Zhu R,
Loeffert JE, Watkins SC and Graham SH: Suppression of endogenous
bcl-2 expression by antisense treatment exacerbates ischemic
neuronal death. J Cereb Blood Flow Metab. 20:1033–1039. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
MacManus JP and Linnik MD: Gene expression
induced by cerebral ischemia: an apoptotic perspective. J Cereb
Blood Flow Metab. 17:815–832. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han B, Wang Q, Cui G, Shen X and Zhu Z:
Post-treatment of Bax-inhibiting peptide reduces neuronal death and
behavioral deficits following global cerebral ischemia. Neurochem
Int. 58:224–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xi HJ, Zhang TH, Tao T, Song CY, Lu SJ,
Cui XG and Yue ZY: Propofol improved neurobehavioral outcome of
cerebral ischemia-reperfusion rats by regulating Bcl-2 and Bax
expression. Brain Res. 1410:24–32. 2011. View Article : Google Scholar : PubMed/NCBI
|