Introduction
Amyotrophic lateral sclerosis (ALS) is a lethal
neurodegenerative disease characterized by the progressive loss of
motor neurons in the motor cortex, brainstem and spinal cord
(1). The clinical manifestations
include the progressive development of major muscle weakness,
atrophy, tremors, tendon reflexes hyperfunction and pathologically
positive features. Approximately 90–95% of all ALS cases are
sporadic and 5–10% of cases are familial (fALS). At present, the
pathogenesis of ALS remains to be fully elucidated and there are no
effective treatment methods. The majority of ALS patients succumb
to respiratory failure within 3–5 years of clinical onset (2).
Present evidence suggests (3,4) that
the progressive loss of motor neurons in ALS cases results from a
series of complex interaction mechanisms, including oxidative
stress, excitotoxicity, altered axonal transport, mitochondrial
dysfunction, an abnormal cytoskeleton, protein aggregation and
genetic factors. In these mechanisms, oxidative stress is
considered to be important in the pathogenesis of selective motor
neuron degeneration and corticospinal tract degeneration (5), and it is also a major contributory
factor leading to chronic motor neuron degeneration and death
(6). Several previous studies have
demonstrated that oxidative stress is present in cerebrospinal
fluid and spinal cord slices of ALS patients, suggesting that
oxidative stress may be a crucial link in the pathogenesis of ALS
(5). In mutant superoxide
dismutase 1 (SOD1) ALS mouse models, the level of oxidative damage
to biological macromolecules (including proteins, lipids and
nucleic acid) was elevated in the brain and spinal cord. Numerous
previous studies have revealed that ~20% of cases of fALS are
caused by mutations in the antioxidant enzyme Cu/Zn SOD1 (7). Rodents, which overexpress mutant
forms of hSOD1, usually develop an ALS-like phenotype (8,9). At
present, the toxic pathogenic mechanisms for human SOD1 mutant
transgenic animals remain unknown. A previous study found that
nuclear factor erythroid 2-related factor 2 (Nrf2) was able to
interact with the antioxidant response element (ARE) to regulate
the expression of phase II antioxidant enzymes including NAD(P)H:
quinone oxidoreductase 1 (NQO1) and heme oxygenase-1 (HO-1). The
Nrf2/ARE signaling pathway is one of the most important endogenous
antioxidant stress pathways (10).
Previous studies have demonstrated that the activation of the
Nrf2/ARE signaling pathway was able to decrease different types of
cellular damage in different tissues and organs (11–13).
At present, the role of the Nrf2/ARE signaling pathway in the onset
of ALS motor neuron degeneration remains to be fully elucidated. In
the present study, the effect of the mutant human SOD1-G93A gene on
the Nrf2/ARE endogenous antioxidant pathway was observed using
NSC-34 cells transfected with human SOD1-G93A, which was already
established in our laboratory. The present study aimed to provide a
further theoretical basis for the pathogenesis and treatment of
ALS.
Materials and methods
Chemicals
An antibody recognizing neurofilament (SMI-32) was
purchased from Covance Inc. (Princeton, NJ, USA). Other antibodies,
including anti-β-actin, anti-Nrf2, anti-NQO1 and anti-HO-1 were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). A First Strand cDNA Synthesis kit was purchased from Thermo
Fisher Scientific (Waltham, MA, USA). A malondialdehyde (MDA) assay
kit was purchased from Nanjing Jiancheng Bioengineering Institute
(Nanjing, Jiangsu, China). A total protein extraction kit was
purchased from Nanjing KeyGen Biotech., Co., Ltd (Nanjing, Jiangsu,
China).
Cell lines and cell cultures
NSC-34 is a hybrid cell line, produced by fusion of
motor neuron enriched, embryonic mouse spinal cord cells with mouse
neuroblastoma, which possesses several of the unique morphological
and physiological characteristics of motor neurons. NSC-34 cells
stably transfected with the empty pcDNA3.1(−) plasmid and the
pcDNA3.1(−) plasmid carrying hSOD1-WT and hSOD1-G93A were
successfully established. The cell lines were removed from liquid
nitrogen and thawed quickly in a 37°C water bath. Then, the cells
were diluted 5-fold with complete medium (containing 90% Dulbecco’s
modified Eagle’s medium, 10% fetal bovine serum, 100 IU/ml
penicillin and 0.1 mg/ml streptomycin) and seeded to 25
cm2 glass culture bottles. The cultures were maintained
at 37°C in a 5% CO2 humidified atmosphere. Following
attachment of the cells to the culture bottles (~4–6 h), the medium
was refreshed to remove the dimethyl sulfoxide. The medium was
generally changed every 2–3 days, depending on the rate of
growth.
Immunocytochemical analysis
An immunocytochemical staining technique was used to
observe the morphology of stably transfected cells. The stably
transfected cells were digested and collected by centrifugation and
then resuspended with culture medium. Then, the cells were seeded
in a 6-well culture plate at a density of 2×104/ml.
Following 24 h, the cells were rinsed with 0.01 M
phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde
for 40 min. To inhibit non-specific binding, the cells were
incubated with goat serum incubated for 15 min. The cultures were
incubated with an anti-neurofilament antibody (SMI-32) overnight at
4°C. The cultures were washed three times with PBS and incubated
with a biotinylated secondary antibody for 15 min in 37°C and then
visualized with 3,3′-diaminobenzidine tetrahydrochloride.
Measurement of MDA
MDA is one of the most important degradation
products of lipid peroxidation. It is able to react with
thiobarbituric acid to generate mauve material. The enzyme activity
was determined by monitoring the change in absorbance at 532
nm.
Reverse transcription polymerase chain
reaction
Total RNA was isolated using TRIzol reagent. The
concentrations and qualities of RNA were determined by measuring
absorbance at 260 and 280 nm. The synthesis of the first chain cDNA
was performed according to the manufacturer’s instructions of the
First Strand cDNA Synthesis kit (Thermo Fisher Scientific). A 25 μl
system of PCR amplification was used and consisted of 12.5 μl of
DreamTaq Green PCR Master mix (2X), 10.5 μl of sterile ultrapure
water, 0.5 μl of cDNA samples and 0.5 μl of forward and reverse
primers. The specific primers used were as follows: β-actin,
forward 5′-GGGACCTGACTGACTACCTCA-3′ and reverse
5′-GACTCGTCATACTCCTGCTTG-3′; Nrf2, forward
5′-ATCGACAGTGCTCCTATGCGTGAA-3′ and reverse
5′-ATCGTCTGGGCGGCGACTTTAT-3′; HO-1, forward
5′-ATCGTGGTGATGGCTTCCTTGT-3′ and reverse
5′-ATCGACCTCGTGGAGACGCTTT-3′; NQO1, forward
5′-ATCGGAGAAGAGCCCTGATTGT-3′ and reverse
5′-ATCGAAAGGACCGTTGTCGTAC-3′. For PCR, the amplification conditions
consisted of an initial activation step at 95°C for 4 min and 30
cycles at 95°C for 15 sec, 60°C for 15 sec, 72°C for 30 sec and
finally 72°C for 7 min. In order to detect the reverse
transcription PCR amplification products, 1.5% agarose gel
electrophoresis was used.
Western blot analysis
The proteins were extracted using a total protein
extraction kit. The extraction of protein was quantified using the
bradford method. A total of 60 mg of extracted protein was resolved
by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
and the resolved proteins in the gel were transferred onto
polyvinylidene difluoride membranes. The membranes were incubated
overnight at 4°C with the following specific primary antibodies:
rabbit polyclonal anti-Nrf2 (1:200), rabbit polyclonal anti-HO-1
(1:200), goat polyclonal anti-NQO1 (1:200) and mouse monoclonal
anti-β-actin (1:500). Then, the membranes were incubated with
corresponding secondary antibody (the dilution of β-actin secondary
antibody was 1:10,000 and the others were 1:3,000) for 1 h at room
temperature and immunodetection was performed with an enhanced
chemiluminescent substrate. The data calculated were obtained by
the rate of the density of target protein banding to the density of
corresponding β-actin banding.
Statistical analysis
The results are expressed as the mean ± standard
deviation. Statistical analyses were performed using two-way
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of the human SOD1-G93A gene on
NSC-34 cellular morphology
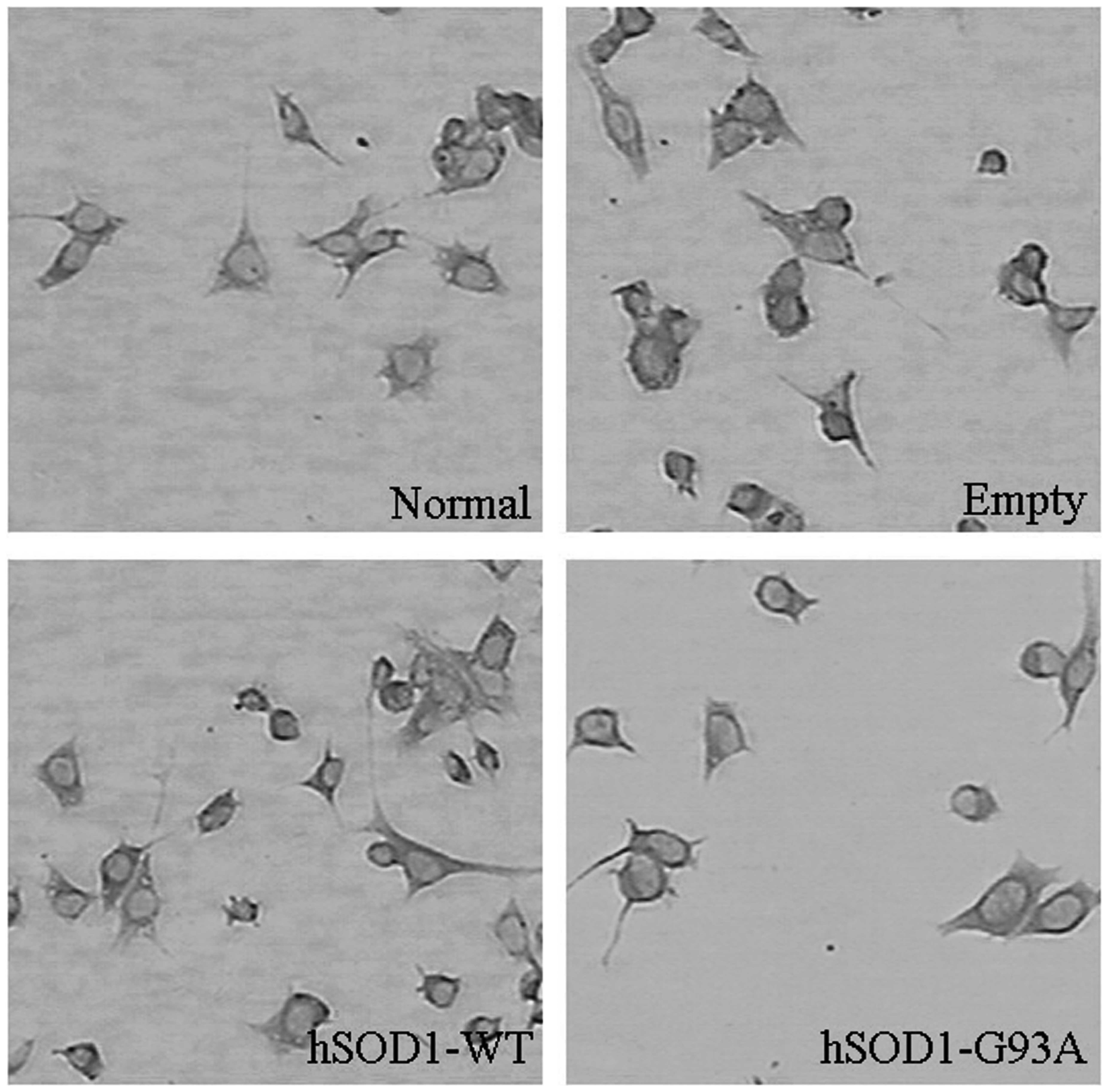
In order to observe the morphological alterations of
cells stably transfected with the pcDNA3.1(−) plasmid, the
hSOD1-pcDNA3.1(−) plasmid and the hSOD1-G93A-pcDNA3.1(-) plasmid,
the stably transfected cells were dyed and examined using the
immunocytochemical method. Observations under the microscope
revealed that the soma exhibited a rounded morphology and the
number of neurites decreased in the NSC-34 cells transfected with
the hSOD1-G93A gene. In addition, the neurites were shorter
compared with the normal NSC-34 cells. However, no significant
changes in the cells transfected with the pcDNA3.1(−) and the
hSOD1-pcDNA3.1(−) plasmid were identified (Fig. 1).
Detection of the lipid peroxidation
level
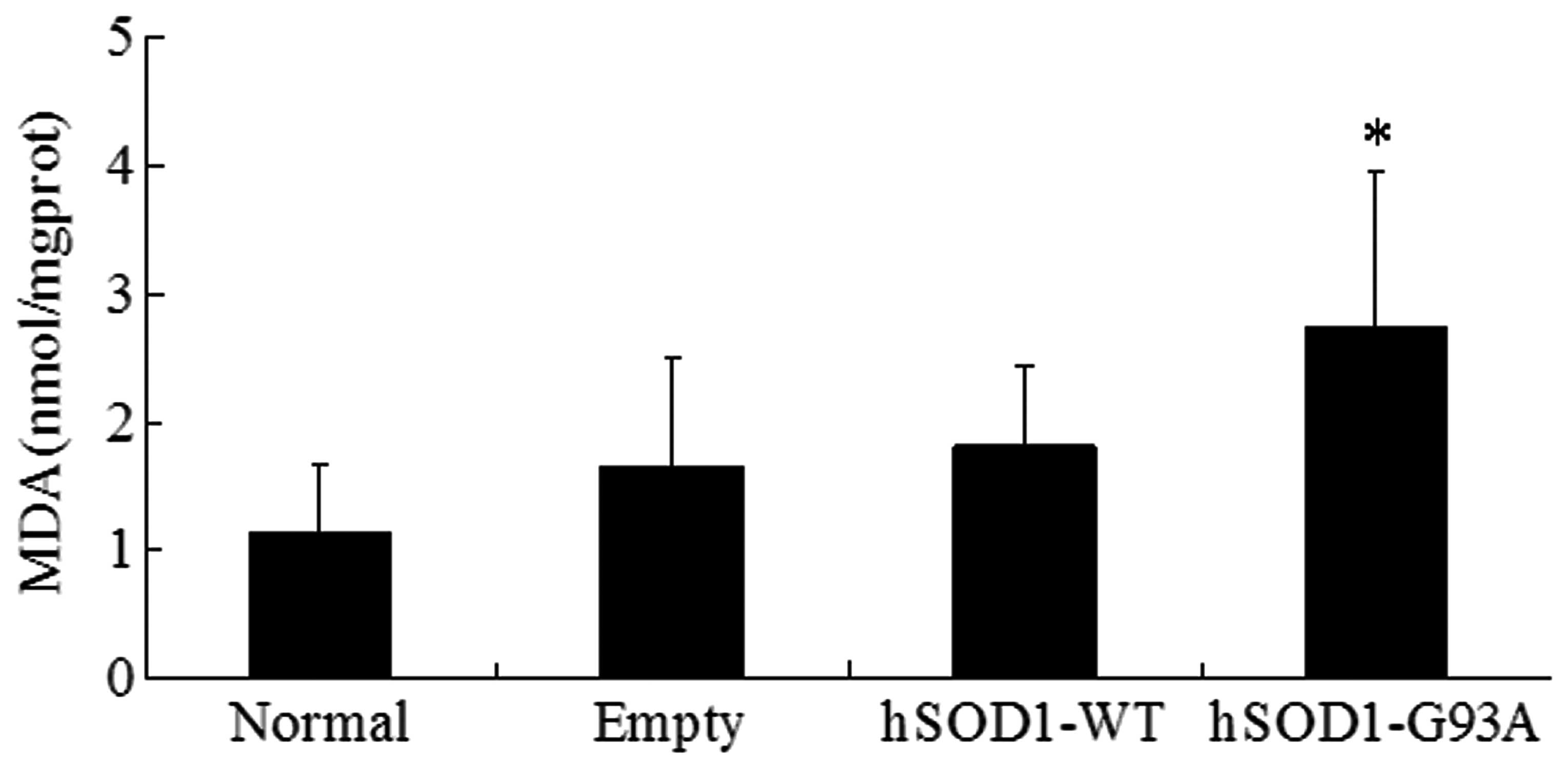
MDA is able to react with thiobarbituric acid to
generate mauve material. The content of MDA directly reflects the
level of lipid peroxidation in cells. The present study found that
the content of MDA was 2.74±0.14 nmol/mg protein in NSC-34 cells
which were stably transfected with the hSOD1-G93A gene. The content
of MDA in the normal, empty and hSOD1-WT cells was 1.15±0.53,
1.65±0.21 and 1.81±0.18 nmol/mg prot, respectively. Statistical
analysis demonstrated that the content of MDA in NSC-34 cells
transfected with the hSOD1-G93A gene was significantly increased
compared with the other three cell lines, while no significant
difference in MDA content in the other three cell lines was
observed (Fig. 2).
Effect of the hSOD1-G93A gene on the mRNA
expression of Nrf2 and phase II antioxidant enzymes in NSC-34
cells
Previous studies and the present study revealed that
the level of oxidative stress in NSC-34 cells transfected with the
hSOD1-G93A gene significantly increased. In order to examine
whether increasing oxidative stress was associated with the effect
of hSOD1-G93A on the Nrf2/ARE signaling pathway, the difference in
the mRNA expression of Nrf2 and phase II enzymes was detected in
four types of cell lines. The mRNA expression of Nrf2 in NSC-34
cells transfected with the hSOD1-G93A gene markedly decreased
compared with the other three cell lines (Fig. 3A and B). Further studies also
revealed that the mRNA expression of the downstream factors HO-1
and NQO1 decreased (Fig. 3A, C and
D). By contrast, no significant differences in the cells
transfected with pcDNA3.1 (−) and hSOD1-WT compared with the normal
NSC-34 cells were identified. These results demonstrated that the
Nrf2/ARE signaling pathway was possibly affected by hSOD1-G93A.
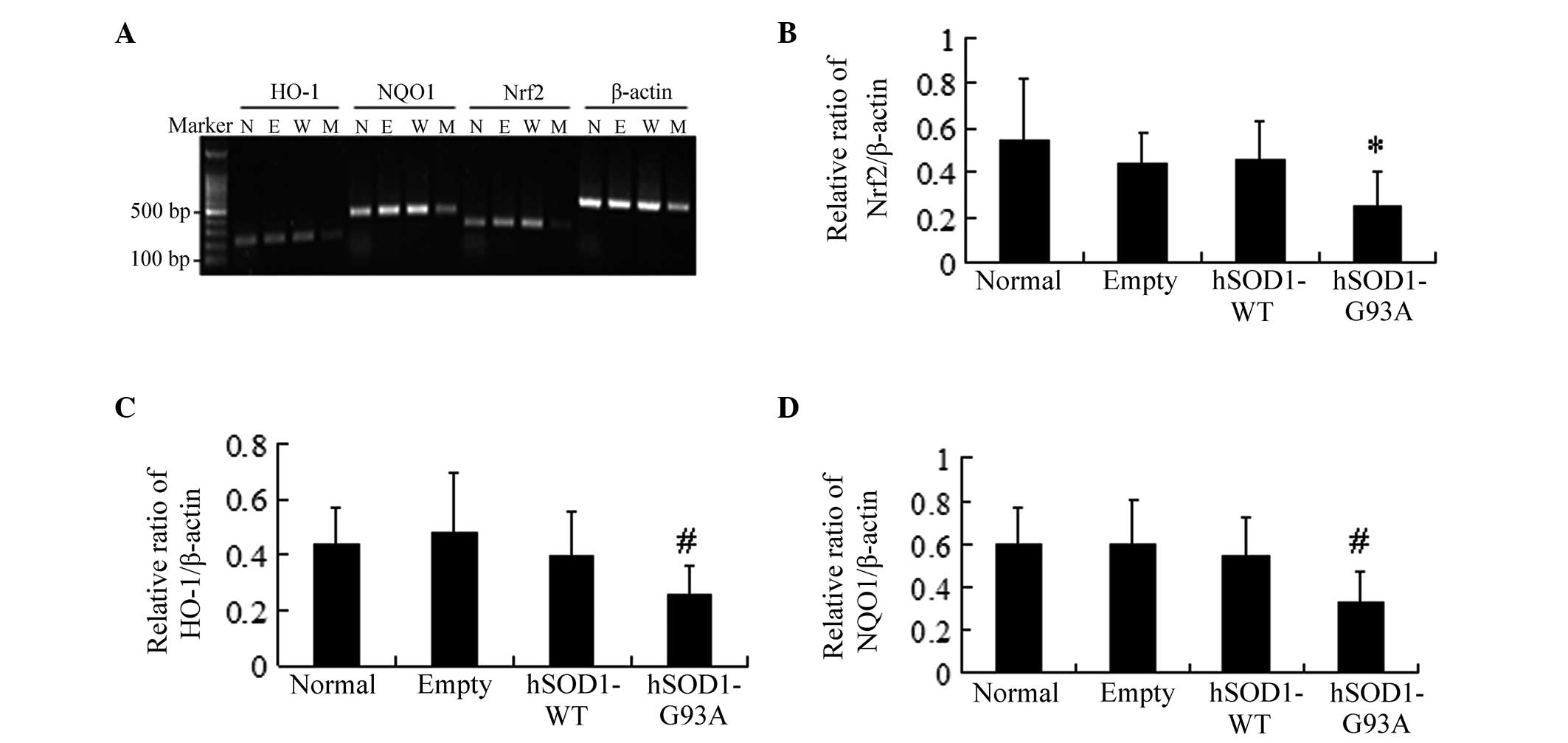 | Figure 3Analysis of the mRNA expression of
Nrf2 and antioxidant response element-driven genes in four cell
lines. (A) Total RNA was extracted from the four cell lines and
HO-1, NQO1 and Nrf2 mRNA levels were determined by PCR and
corrected by β-actin mRNA levels (N, normal: E, empty; W, hSOD1-WT;
M, hSOD1-G93A). (B) Densities of Nrf2 bands were measured and its
ratio to β-actin was calculated (mean ± standard deviation; n=6).
(C) Densities of HO-1 bands were measured and its ratio to β-actin
was calculated (mean ± standard deviation; n=8). (D) Densities of
NQO1 bands were measured and its ratio to β-actin was calculated
(mean ± standard deviation; n=6). *P<0.05 and
#P<0.01, statistically different from the normal,
empty, hSOD1-WT cell lines. Nrf2, nuclear factor erythroid
2-related factor 2; NQO1, NAD(P)H: quinone oxidoreductase 1; HO-1,
heme oxygenase-1; WT, wild type; hSOD1, human superoxide dismutase
1. |
Effect of the hSOD1-G93A gene on the
protein expression of Nrf2 and phase II antioxidant enzymes in
NSC-34 cells
In order to further observe the effect of hSOD1-G93A
on the Nrf2/ARE signaling pathway in NSC-34 cells, the differences
in the protein expression of Nrf2 and phase II enzymes in these
four types of cell lines were detected. The western blots
demonstrated that the protein expression of Nrf2 in NSC-34 cells
transfected with the hSOD1-G93A gene markedly decreased compared
with the other three cell lines (Fig.
4A and B; P<0.01). Furthermore, the protein expression of
the downstream factors HO-1 and NQO1 decreased as well (Fig. 4C–F; P<0.01). However, no
significant difference in the protein expression of Nrf2, HO-1 and
NQO1 in the other three cell lines was identified. This phenomenon
was consistent with the results from the genetic test.
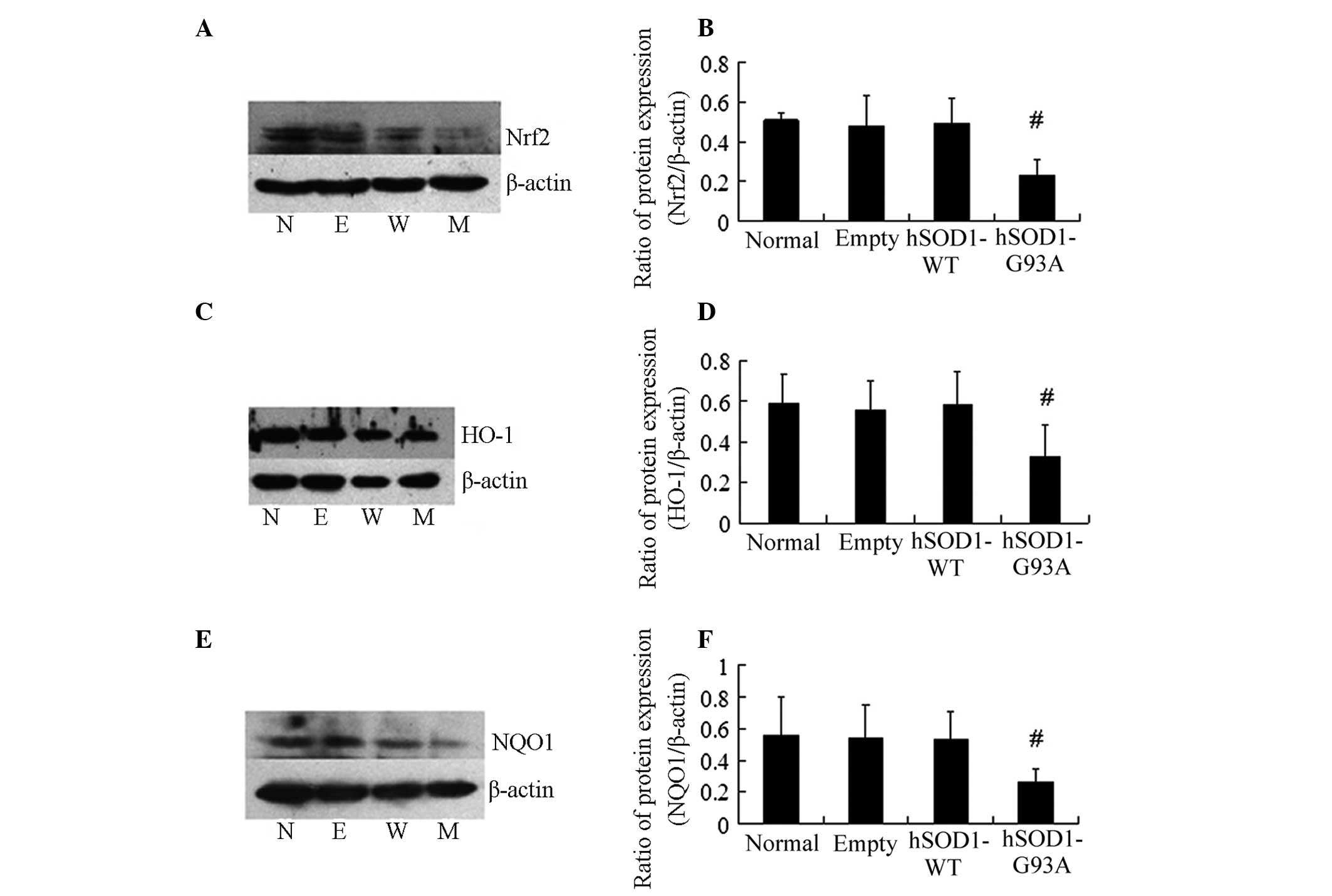 | Figure 4Western blot analysis to measure the
protein expression levels of HO-1, NQO1 and Nrf2 in four cell
lines. (A and B) Densities of Nrf2 bands were measured and its
ratio to β-actin was calculated (mean ± standard deviation; n=4; N,
normal; E, empty; W, hSOD1-WT; M, hSOD1-G93A). (C and D) Densities
of HO-1 bands were measured and its ratio to β-actin was calculated
(mean ± standard deviation; n=6). (E and F) Densities of NQO1 bands
were measured and its ratio to β-actin was calculated (mean ±
standard deviation; n=6). #P<0.01, statistically
different from the normal, empty, hSOD1-WT cell lines. Nrf2,
nuclear factor erythroid 2-related factor 2; NQO1, NAD(P)H: quinone
oxidoreductase 1; HO-1, heme oxygenase-1; WT, wild type; hSOD1,
human superoxide dismutase 1. |
Discussion
In order to investigate the toxic effect of
hSOD1-G93A on motor neurons, the NSC-34 cell lines were selected
and the effect of the hSOD1-G93A gene on the Nrf2/ARE endogenous
antioxidant pathway was observed. NSC-34 cells transfected with
hSOD1-G93A were already established and commonly used in our
laboratory. The present study found that the soma exhibited a
rounded morphology and the number of neurites decreased in the
NSC-34 cells transfected with the hSOD1-G93A gene. In addition, the
neurites were shorter compared with the normal NSC-34 cells.
Furthermore, the hSOD1-G93A gene reduced the ability of cells to
protect against oxidative damage and increased the levels of lipid
peroxidation in cells. Oxidative damage has been substantiated by
the biochemical and histopathological study in postmortem tissue of
ALS patients (14,15). These studies revealed that the
levels of lipid peroxidation, nucleic acid oxidation and protein
nitration in ALS patients significantly increased. In order to
observe the degree of oxidative damage, the intracellular MDA
levels in normal, empty, hSOD1-WT and hSOD1-G93A cell lines were
detected. The results from the present study demonstrated that the
intracellular MDA levels in hSOD1-G93A cell lines significantly
increased compared with the other three cell lines (Fig. 2). This result indicated that the
activity of antioxidants and detoxification of the cell lines
transfected with the hSOD1-G93A gene decreased.
Furthermore, the present study found that the mRNA
expression of Nrf2 in NSC-34 cells, which was stably transfected
with the hSOD1-G93A gene, markedly decreased compared with the
other three cell lines (Fig. 3A and
B). In addition, the mRNA expression of the downstream factors
HO-1 and NQO1 also decreased (Fig. 3A,
C and D). To further investigate the effect of hSOD1-G93A on
the Nrf2/ARE signaling pathway in cells, the western blots of total
cellular protein indicated that the protein expression of Nrf2,
HO-1 and NQO1 significantly decreased in the mutated hSOD1 NSC-34
cell lines (Fig. 4). This is
consistent with the result obtained by Sarlette et al
(16). The authors reported that
the protein expression of Nrf2 was reduced in neurons from the
primary motor cortex and spinal cord from ALS postmortem tissue
samples. In addition, the level of mRNA encoding Nrf2 was decreased
in embryonic motor neurons isolated from hSOD1-G93A rats (17). Petri et al (6) hypothesized that the decrease in the
protein expression of Nrf2 in motor neurons was possibly associated
with the decline in the antioxidant defense mechanism of the body.
Under basal conditions, the Nrf2 was usually bound to the
endogenous inhibitor Kelch-like ECH associated protein 1 (Keap1) in
the cell plasma and was in the inactive state. However, when the
cells were exposed to oxidative stress, Nrf2 was able to dissociate
from the Keap1 protein and translocate to the nucleus, and then
bind to ARE and activate the expression of target genes, including
HO-1 and NQO1 (18,19). Since the cells demonstrated a high
vulnerability to oxidative damage in the HO-1−/− mouse model, HO-1
was considered to be important in the process of endogenous defense
against oxidative stress (20).
Certain studies suggested that NQO1 was directly able to protect
tissues against oxidative stress (21). In cells, NQO1 was a type of
electronic reductase regulating substances in the redox state and
was able to catalyze the reduction reaction of Quinones and its
derivatives. The characteristics of the reaction process did not
generate these products, including free radical oxidation and half
quinone, which is a single electron reductase. Therefore, the
process had a protective function against oxidative stress caused
by metabolism. The current study suggested that destroying the Nrf2
molecules and its downstream signaling pathway was able to
aggravate the deterioration of oxidative damage, inflammation and
mitochondrial dysfunction in cells (22). Furthermore, several previous
studies (17,23) indicated that activated Nrf2 was
beneficial to organisms in the context of hSOD1-G93A toxicity, and
selective Nrf2 overexpression in neurons or type II skeletal muscle
fibers was able to delay disease onset in hSOD1-G93A mice. Various
previous studies revealed that phase II enzyme inducers were able
to upregulate the expression of HO-1 and protect neurons from the
damage caused by oxidative stress through Nrf2/ARE pathways
(24,25). The above evidence suggested that
the Nrf2/ARE signaling pathway is able to protect against oxidative
stress damage and subsequently protect the neurons in the ALS cell
model. Therefore, in the present study, the cause of increased
oxidative stress levels in the mutant hSOD1 of NSC-34 cells may be
associated with the result that the human SOD1-G93A gene affected
the Nrf2/ARE signaling pathway and reduced the expression of HO-1
and NQO1 in NSC-34 cells. In addition, these findings suggest that
activating the Nrf2/ARE signaling pathway and increasing phase II
enzymes are potential therapeutic strategies for the treatment of
ALS.
In conclusion, the present study demonstrated that
the human SOD1-G93A gene damaged the Nrf2/ARE signaling pathway,
reduced the ability of cells to protect against oxidative damage
and increased the vulnerability of motor neurons exposed to
oxidative stress.
Acknowledgements
This study was supported in part by a grant from the
Natural Science Foundation of Shandong, China (grant no.
ZR2010HM041) and a grant from the Post-doctoral Innovation
Foundation of Shandong, China (grant no. 201102004).
References
|
1
|
Avossa D, Grandolfo M, Mazzarol F, et al:
Early signs of motoneuron vulnerability in a disease model system:
characterization of transverse slice cultures of spinal cord
isolated from embryonic ALS mice. Neuroscience. 138:1179–1194.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mitchell JD, Callagher P, Gardham J, et
al: Timelines in the diagnostic evaluation of people with suspected
amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) - a
20-year review: can we do better? Amyotroph Lateral Scler.
11:537–541. 2010.PubMed/NCBI
|
|
3
|
Kiernan MC, Vucic S, Cheah BC, et al:
Amyotrophic lateral sclerosis. Lancet. 377:942–955. 2011.
View Article : Google Scholar
|
|
4
|
Zinman L and Cudkowicz M: Emerging targets
and treatments in amyotrophic lateral sclerosis. Lancet Neurology.
10:481–490. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barber SC and Shaw PJ: Oxidative stress in
ALS: key role in motor neuron injury and therapeutic target. Free
Radical Biol Med. 48:629–641. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Petri S, Körner S and Kiaei M: Nrf2/ARE
signaling pathway: key mediator in oxidative stress and potential
therapeutic target in ALS. Neurol Res Int. 2012:8780302012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valdmanis PN, Daoud H, Dion PA and Rouleau
GA: Recent advances in the genetics of amyotrophic lateral
sclerosis. Curr Neurol Neurosci Rep. 9:198–205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Howland DS, Liu J, She Y, et al: Focal
loss of the glutamate transporter EAAT2 in a transgenic rat model
of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc
Natl Acad Sci USA. 99:1604–1609. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gurney ME, Pu H, Chiu AY, et al: Motor
neuron degeneration in mice that express a human Cu,Zn superoxide
dismutase mutation. Science. 264:1772–1775. 1994. View Article : Google Scholar
|
|
10
|
Yu X and Kensler T: Nrf2 as a target for
cancer chemoprevention. Mutat Res. 591:93–102. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Taguchi K, Motohashi H and Yamamoto M:
Molecular mechanisms of the Keap1-Nrf2 pathway in stress response
and cancer evolution. Genes Cells. 16:123–140. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vargas MR and Johnson JA: The Nrf2-ARE
cytoprotective pathway in astrocytes. Expert Rev Mol Med.
11:e172009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hybertson BM, Gao B, Bose SK and McCord
JM: Oxidative stress in health and disease: the therapeutic
potential of Nrf2 activation. Mol Aspects Med. 32:234–246. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mariani E, Polidori MC, Cherubini A and
Mecocci P: Oxidative stress in brain aging, neurodegenerative and
vascular diseases: an overview. J Chromatogr B Analyt Technol
Biomed Life Sci. 827:65–75. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shibata N, Nagai R, Uchida K, et al:
Morphological evidence for lipid peroxidation and protein
glycoxidation in spinal cords from sporadic amyotrophic lateral
sclerosis patients. Brain Res. 917:97–104. 2001. View Article : Google Scholar
|
|
16
|
Sarlette A, Krampfl K, Grothe C, et al:
Nuclear erythroid 2-related factor 2-antioxidative response element
signaling pathway in motor cortex and spinal cord in amyotrophic
lateral sclerosis. J Neuropathol Exp Neurol. 67:1055–1062. 2008.
View Article : Google Scholar
|
|
17
|
Pehar M, Vargas MR, Robinson KM, et al:
Mitochondrial superoxide production and nuclear factor erythroid
2-related factor 2 activation in p75 neurotrophin receptor-induced
motor neuron apoptosis. J Neurosci. 27:7777–7785. 2007. View Article : Google Scholar
|
|
18
|
Neymotin A, Calingasan NY, Wille E, et al:
Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and
CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral
sclerosis. Free Radic Biol Med. 51:88–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Malhotra D, Portales-Casamar E, Singh A,
et al: Global mapping of binding sites for Nrf2 identifies novel
targets in cell survival response through ChIP-Seq profiling and
network analysis. Nucleic Acids Res. 38:5718–5734. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Poss KD and Tonegawa S: Reduced stress
defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci
USA. 94:10925–10930. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Siegel D, Gustafson DL, Dehn DL, et al:
NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger.
Mol Pharmacol. 65:1238–1247. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Slocum SL and Kensler TW: Nrf2: control of
sensitivity to carcinogens. Arch Toxicol. 85:273–284. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vargas MR, Burton NC, Kutzke J, et al:
Absence of Nrf2 or its selective overexpression in neurons and
muscle does not affect survival in ALS-linked mutant hSOD1 mouse
models. PLoS One. 8:e566252013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun MM, Bu H, Li B, et al: Neuroprotective
potential of phase II enzyme inducer diallyl trisulfide. Neurol
Res. 31:23–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stack C, Ho D, Wille E, et al:
Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide
improve the behavioral phenotype and brain pathology in a
transgenic mouse model of Huntington’s disease. Free Radic Biol
Med. 49:147–158. 2010.PubMed/NCBI
|