Introduction
Osteoarthritis (OA) is the most prevalent type of
joint disease in adults and is characterized by chronic pain and
degradation of articular cartilage (1,2).
Treatments for early stage OA initially focus on reducing pain and
inflammation, and preventing the cartilage from further mechanical
injury. Glucocorticosteroids (GCs) are intra-aritcular therapeutics
first used in for anti-inflammatory treatment of rheumatoid
arthritis in 1951 and are effective in pain relief and functional
improvement (3–6). However, long-term and/or high-dose
treatment with GCs may be harmful for articular cartilage
metabolism and may induce apoptosis in chondrocytes by destroying
the endoplasmic reticulum and mitochondria (7,8). In
cartilage, chondrocytes are the sole resident cells that are
involved in the synthesis of the extracellular matrix (ECM).
Cartilage homeostasis requires sufficient ECM composition to exert
its biomechanical functions. Therefore, inhibition of the growth of
chondrocytes may reduce cartilage formation and possibly weaken the
matrix structure.
Autophagy is characterized by the formation of an
autophagosome. As the essential cellular homeostasis mechanism, the
autophagosome may recycle the damaged and dysfunctional organelles
and molecules (9,10). Therefore, autophagy is considered a
protective process for cell survival under metabolic stress and
adverse stimulus (11). Thus,
impairment of autophagy may induce a variety of human diseases. Due
to the relatively low rate of proliferation of chondrocytes,
autophagy appears to be important in maintaining cell survival and
biosynthetic function (12,13).
However, whether the interaction between autophagy and apoptosis is
involved in the effect of GC on normal human chondrocytes remains
to be elucidated.
Rapamycin (Rapa) is a lipophilic macrolide
antibiotic, which triggers autophagic functions by inhibiting the
mammalian target of rapamycin (mTOR) (14). mTOR, a nutrient-sensing kinase
which acts as a controller of ribosomal biogenesis and protein
synthesis, may directly suppress cell autophagy (15). In addition, imbalances of this
kinase are involved in metabolic diseases (16). Based on the potential of inducing
autophagy, Rapa has been suggested to protect against certain
degenerative conditions (17–19).
Recently, a study by our group demonstrated that
short-term GC treatment did not suppress chondrocyte growth
significantly, whereas long-term exposure to GCs induced apoptosis
(20). Thus, the aim of the
present study was to determine whether autophagy is one of the
survival pathways through which chondrocytes respond to short-term
GC treatment and whether pharmacological enhancement of autophagy
by Rapa may serve as a protective mechanism against the adverse
effects of long-term GC exposure.
Materials and methods
Chemicals and reagents
Dexamethasone (Dex), Rapa, monodansylcadaverine
(MDC), Hoechst 33342 and dimethylsulfoxide (DMSO) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). A CCK-8 Cell Counting Kit
was obtained from Beyotime Laboratories (Beyotime Institute of
Biotechnology, Haimen, China). Annexin V-fluorescein isothiocyanate
(FITC) and propidium iodide (PI) were purchased from BaoSai
Biotechnology Co., Ltd. (Beijing, China). Antibodies against
microtubule-associated protein light chain 3 (LC3), beclin-1,
caspase-9, poly (adenosine diphosphate-ribose) polymerase (PARP),
collagenase II, aggrecan and β-actin were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
Human chondrocyte isolation and
culture
Human articular cartilage tissue samples from the
knee joint were obtained from 10 donors (average age, 50.2 years)
undergoing surgery for tibial plateau fracture or total knee
arthroplasty. These surgeries were approved by the ethics committee
of The First Affiliated Hospital of Harbin Medical University
(Harbin, China). The non weight-bearing area of the cartilage,
without any abnormalities, was harvested and washed in sterilized
saline. Next, the tissue was sliced into smaller sections of 1
mm3 and digested with trypsin and collagenase II for 5 h
at 37°C. The mixture was filtered, centrifuged (0.2 × g) and washed
in phosphate-buffered saline (PBS) at least three times for cell
purification. Finally, the cells were transferred into a culture
flask and incubated at 37°C in a 5% CO2 incubator with
10% fetal bovine serum in Dulbecco’s modified Eagle medium (DMEM)
(Gibco-BRL, Carlsbad, CA, USA). Following the above procedure, the
second-passage chondrocytes were used for further experiments.
Cell viability assay
Chondrocytes (8×103) were seeded into
96-well plates and cultured for 24 h. Based on the previous
results, the cells were treated with 100 μM/l Dex in culture medium
for the following times: 6, 12, 24, 48 or 72 h. Cells were treated
with DMSO only as the untreated control (control group), and one
group was pretreated with 10 μM/l Rapa for 2 h. At each time-point,
10 μl CCK-8 kit solution was added to each well and plates were
incubated at 37°C for ~1 h. Next, a microplate reader (Thermo
Fisher Scientific Inc., Walthom, MA, USA) was used to measure the
optical density (OD) at 450 nm. The experiment was repeated at
least five times. The survival rate of cells (%) = (experimental
group OD value - blank group OD value)/(control group OD value -
blank group OD value), and the inhibition rate of cell
proliferation (%) equals to 100% minus the survival rate.
Flow cytometric assessment of
apoptosis
Following the respective treatment as above, the
cells were washed with ice-cold PBS, harvested and counted. The
cells (1×105) were suspended in 500 μl binding buffer
containing Hepes/NaOH (pH 7.4), NaCl and CaCl2 (BaoSai
Biotechnology Co., Ltd.) and incubated with 10 μl Annexin V-FITC
and 5 μl PI for 20 min. The rate of apoptosis (%) was measured by a
flow cytometer (Epics Altra II; Beckman Coulter Inc., Brea, CA,
USA). In addition, for Hoechst staining, the cells were seeded into
6-well plates and cultured for 24 h. After treatment with or
without 100 μM of Dex for 72 h post-treatment, the cells were
washed in cold PBS twice and were labeled with Hoechst 33258
staining solution (Beyotime Institute of Biotechnology), and
maintained at 37°C in the dark for 20 min. The cells were then
observed and imaged by fluorescence microscopy (Leica Microsystems,
Wetzlar, Germany), with excitation at 350 nm and emission at 460
nm.
MDC assay
Chondrocytes were seeded on sterile coverslips in
tissue culture plates. Following treatment with Dex and/or Rapa for
the indicated times, the cells were incubated with MDC (0.1 mM) for
30 min at 37°C. Following incubation, cells were washed three times
with PBS and immediately visualized by fluorescence microscopy
(Leica Microsystems). Excitation wavelengths were 360–380 nm and
Olympus DP version software (Olympus Corporation, Tokyo, Japan) was
used.
Transmission electron microscopy (TEM)
analysis
Chondrocytes were treated with Dex and/or for
designated times as described for the MDC assay. Cells were then
washed with 0.1 M cacodylate buffer (pH 7.4) and fixed with 2%
glutaraldehyde in PBS for 24 h at 4°C. Samples were then processed
following the standard procedure (21). A Zeiss transmission electron
microscope (Carl Zeiss, Thornwood, NY, USA) was used to examine the
section samples.
Western blot analysis
Chondrocytes (2×106) were pretreated with
or without Rapa (10 μM/l) 2 h prior to Dex treatment. The cells
were collected and western blot analysis was performed as
previously described (19).
Briefly, the cells were washed with ice-cold PBS and sonicated in
radioimmunoprecipitation assay buffer and homogenized and cellular
debris was removed by centrifugation (0.2 × g). The protein
concentration was determined using the Bio-Rad DC protein assay
(Bio-Rad Laboratories, Hercules, CA, USA). The protein lysates were
separated by 12 or 15% SDS-PAGE and transferred to polyvinylidene
difluoride membranes. The membranes were blocked with 5% bovine
serum albumin (Beyotime Institute of Biotechnology) and then
incubated with LC3 and Beclin-1 primary antibodies (1:500), Cleaved
caspase-9 and PARP primary antibodies (1:1,000) overnight at 4°C
and subsequently with horseradish peroxidase-conjugated anti-rabbit
(1:1,500) or anti-mouse (1:2,000) secondary antibodies. The protein
blots were visualized by enhanced chemiluminescence (Millipore,
Billerica, MA, USA) with Chemilumino Analyzer LAS-3000 (Fujifilm,
Tokyo, Japan). Quantity One 1-D Analysis software (Bio-Rad
Laboratories) was used to analyze the band density of each blot.
Anti β-actin was also used as an internal control and to confirm
that each sample contained a similar quantity of protein.
Statistical analysis
All the experimental data presented were confirmed
in at least three independent experiments, unless otherwise
indicated. The experimental results are expressed as the mean ±
standard deviation. Collected data were statistically analyzed
using a one-way analysis of variance and the Fisher’s
least-significant difference test with SPSS 19.0 software (SPSS
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of GC treatment on the viability
of human chondrocytes
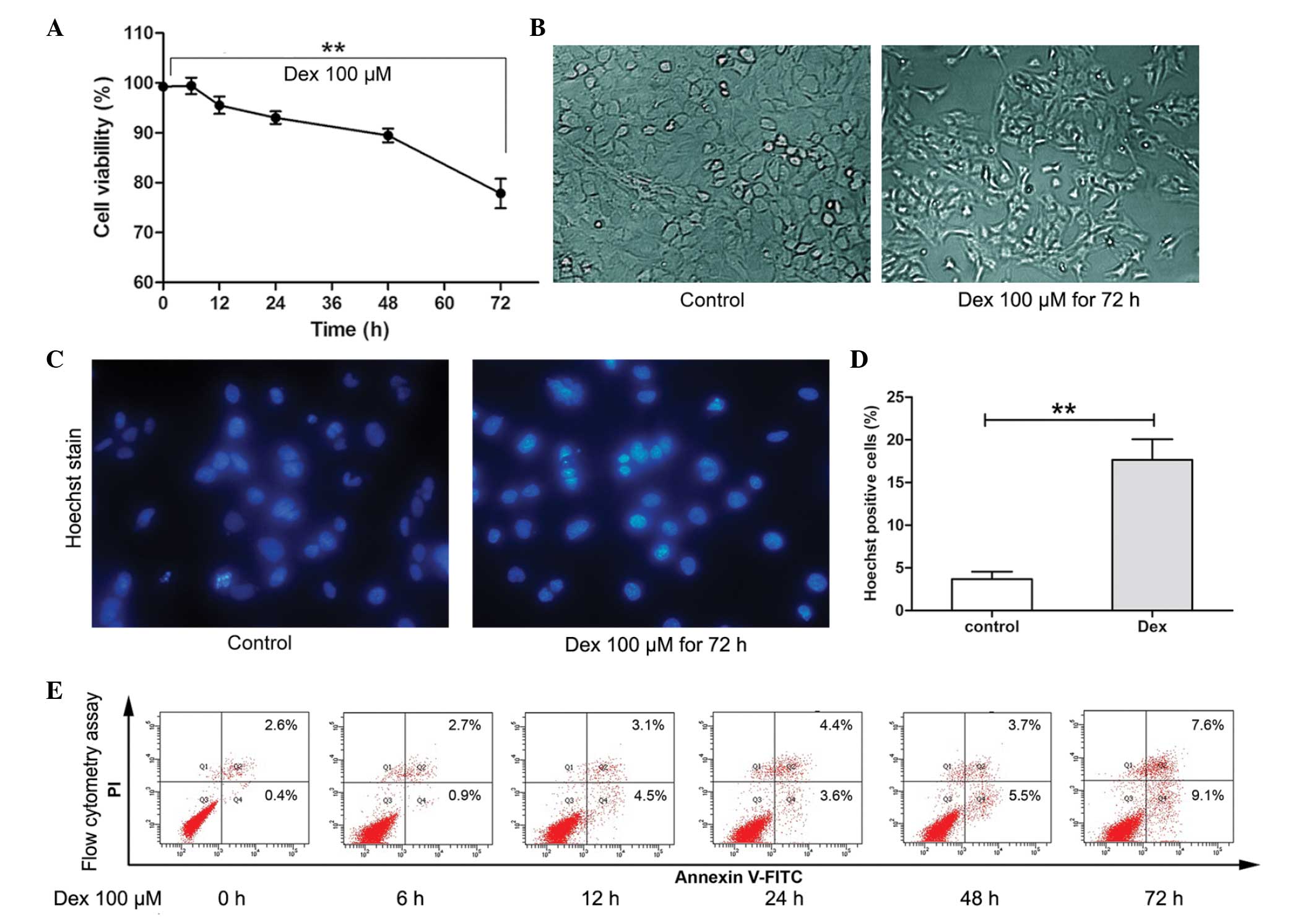
It has been previously observed that, following 72 h
exposure, Dex at 100 μM/l suppressed cell growth significantly
(18). Therefore, in the present
study, the short-term effect of Dex stimulation on the viability of
chondrocytes was determined using the CCK-8 assay. As shown in
Fig. 1A and B, during short-term
treatment (6–24 h), the average viability of chondrocytes remained
>90%. However, following 72 h treatment, the viability of cells
was decreased significantly (P<0.01) and cell morphological
changes were observed. To further determine the cytotoxicity of Dex
on chondrocytes, flow cytometry and Hoechst staining were used to
assess apoptosis. As shown in Fig.
1C–E, following 100 μM/l Dex treatment for 6, 12, 24 and 48 h,
a slight increase in the rate of apoptosis was detected (3.6–9.2%).
However, following treatment for 72 h, the rate of apoptosis
increased to 16.7%. Furthermore, Hoechst 33342 staining showed that
cells treated with Dex for 72 h exhibited an increased ratio of
Hoechst-labeled (bright blue nuclei indicative of apoptosis) cells
compared with untreated cells (**P<0.01). These
results indicated that short-term Dex treatment did not induce
significant levels of apoptosis-associated cell death in
chondrocytes; however, long-term treatment may induce apoptosis
significantly.
Short-term GC treatment induces autophagy
in chondrocytes
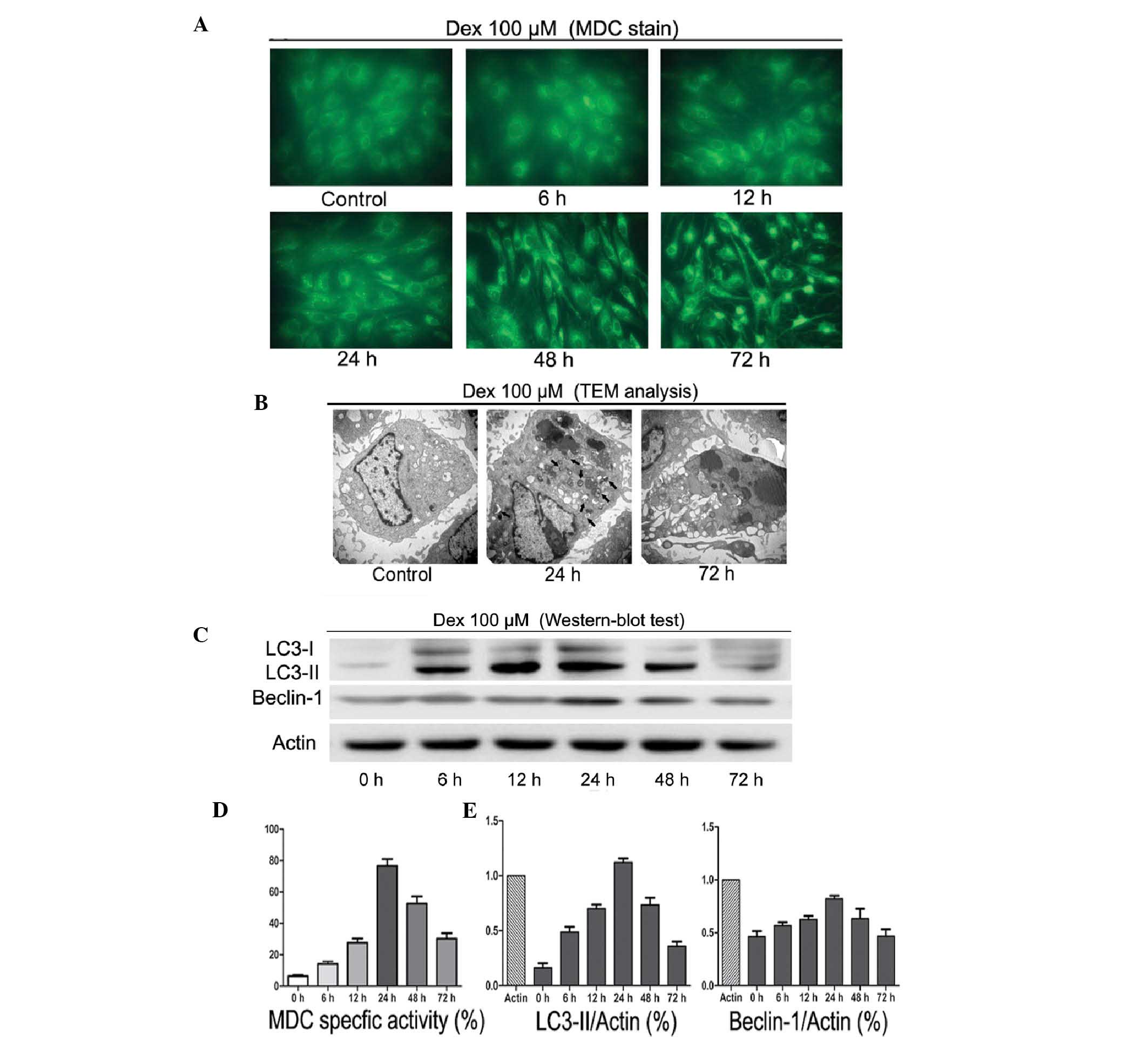
To investigate the autophagic activity following Dex
treatment, MDC staining, TEM and western blot analysis were used.
As shown in Fig. 2A and D, 100
μM/l Dex induced autophagy in a time-dependent manner. Between 6
and 24 h of treatment, MDC-labeled vacuoles were significantly
increased. By contrast, following 48 and 72 h Dex treatment, a
decrease in the occurrence of cell morphological changes and
MDC-labeled vacuoles was observed. TEM scanning showed a large
number of free membrane structures and double-membrane vacuoles,
which contained portions of cytosol and organelles, in the
cytoplasm following treatment with Dex for 24 h as compared with
untreated cells. These structures resembled pre-autophagosomal
structures (PAS) or autophagosomes (Fig. 2B, black arrows scale bars). Western
blot analysis showed that LC3-II and Beclin-1 protein levels
significantly increased between 6 and 24 h of treatment (Fig. 2C and E). However, this trend was
reversed following long-term exposure to Dex. These observations
suggested that short-term treatment with GCs may induce autophagy
in chondrocytes.
Effects of activation of autophagy by
Rapa on apoptosis of chondrocytes
It has been reported that Rapa may induce autophagy
in chondrocytes. To further confirm the protective effect of Rapa
in chondrocyte exposure to GC, the cells were pretreated with Rapa
2 h prior to treatment with Dex. The results suggested that,
compared with Dex only treatment, pre-culture of the cells with
Rapa enhanced the cell viability significantly (P<0.05; Fig. 3A) and reduced the apoptotic rates
(Fig. 3B). Furthermore, the
activity of autophagy was stimulated by Rapa. The number of
MDC-specific vacuoles was significantly increased (P<0.01;
Fig. 3C and D). Finally, LC3-II
protein expression was also upregulated, whereas the expression of
apoptosis-associated gene caspase-9 and the protein PARP were
downregulated (Fig. 3E and F).
These results suggested that the apoptosis induced by Dex may be
through the activation of the caspase signaling pathway and that
Rapa may maintain chondrocyte survival in the presence of GC by
enhancing autophagy and suppressing apoptosis.
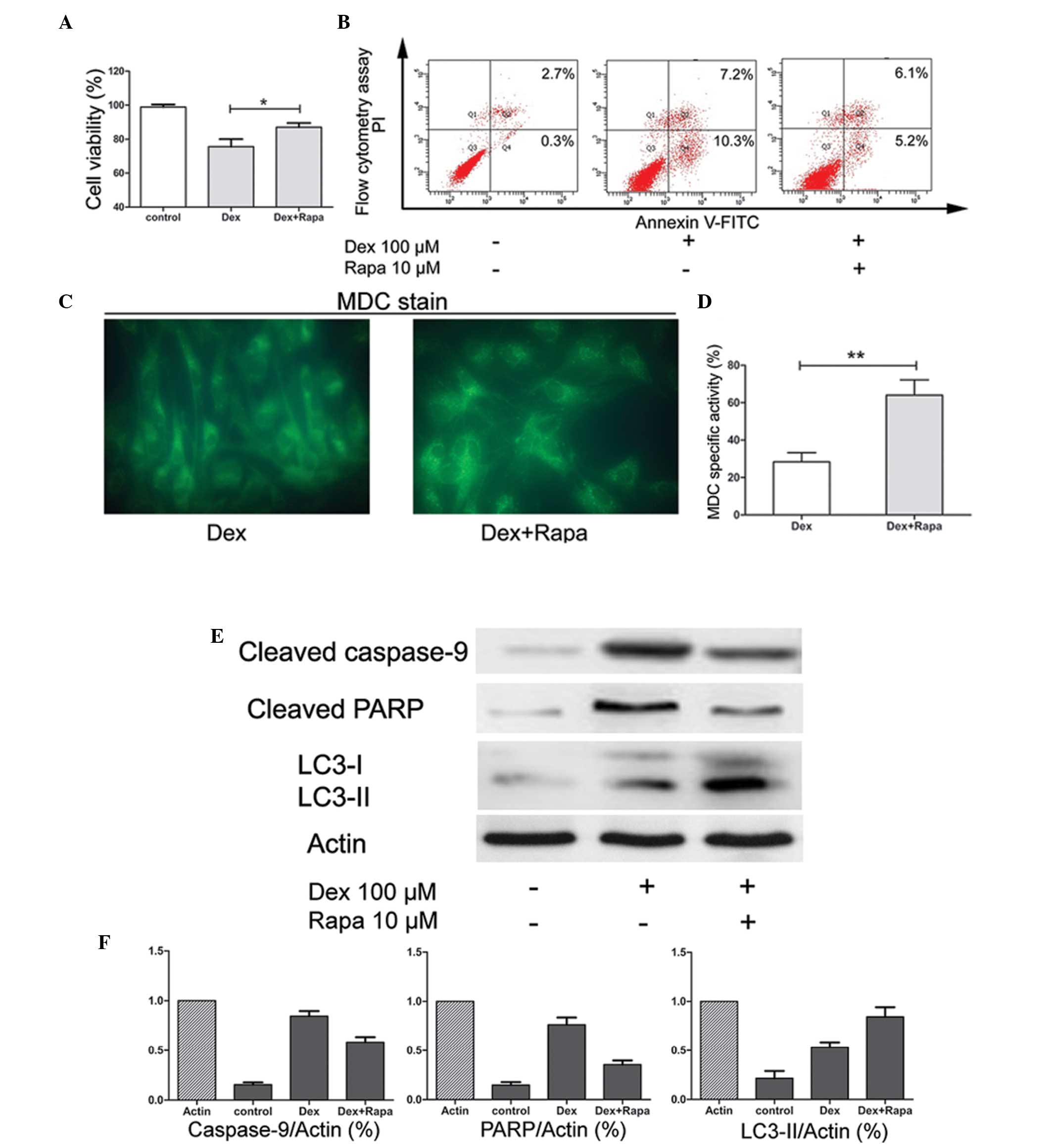 | Figure 3Effects of activation of autophagy by
Rapa. (A) Chondrocytes were pretreated with or without Rapa and
exposed to Dex for 72 h. The cell viability index (%) was obtained
using a CCK-8 assay. Data are expressed as the mean ± SD and
significant differences are indicated by asterisks
(*P<0.05, vs. Dex). (B) Following treatment, the
apoptotic index was determined by flow cytometry following Annexin
V-FITC and PI staining. (C) Following Dex only or pretreatment with
Rapa with subsequent Dex exposure for 72 h, the MDC-labeled
vacuoles in chondrocytes were examined by fluorescence microscopy
(magnification, ×400). (D) The specific activity index (%) was
obtained by flow cytometry. Data are expressed as the mean ± SD and
significant differences are indicated by asterisks
(**P<0.01, vs. Dex). (E) Western blot analysis for
cleaved caspase-9, cleaved PARP, LC3-I and LC3-II protein following
72 h of incubation of untreated samples, Dex-treated samples and
samples pretreated with Rapa followed by Dex treatment. β-actin was
used as a internal control. (F) Levels of proteins were normalized
to the β-actin band density using Bio-Rad Quantity One software.
Data were expressed as the mean ± SD. Rapa, rapamycin; Dex,
dexamethasone; MDC, monodansylcadaverine; FITC, fluorescein
isothiocyanate; SD, standard deviation; PI, propidium iodide; LC3,
microtubule-associated protein light chain 3; PARP, poly (adenosine
diphosphate-ribose) polymerase. |
Discussion
OA is the most common type of joint disease
worldwide, which is mainly characterized by chronic, irreversible
degradation and erosion of cartilage (1,22).
For the majority of patients, joint pain and movement disorders are
the primary clinical symptoms. At early stages, if the cartilage
damage is not serious and joint pain is the major complaint,
nonoperative therapy is preferred by the majority of clinicians
(23). Glucocorticoid drugs,
including Dex and prednisone, are effective anti-inflammatory
agents in intra-articular injection therapy. Numerous studies have
demonstrated the positive effects of GC in the treatment of early
OA (24,25). However, GC treatment alters
cartilage metabolism and changes the intra-articular environment
(7,8,26).
Therefore, a number of studies have hypothesized that the repeated
use of GC may facilitate cartilage degeneration (27,28).
In a recent study, long-term GC treatment was observed to inhibit
the growth of chondrocytes and restrain the synthesis of the
extracellular matrix (20). In the
present study, it was observed that short-term GC treatment did not
significantly reduce chondrocyte viability.
Autophagy, or the autophagic process, is a
well-conserved mechanism among species and has been confirmed to be
important in various biological events (9,10,11,28).
This system is characterized by the formation of double-membrane
autophagic vesicles, which contain cytoplasm, and/or organelle
degradation by lysosomes for material recycling and ATP generation
(29). Among the human autophagy
genes, Beclin-1 and LC3 are major regulators and markers of the
autophagy pathway (30). Beclin-1
forms a complex with type III phosphatidylinositol that allows
nucleation of the autophagic vesicles. During the process of
autophagy activation, LC3-I is converted into LC3-II, which is then
attached to the membrane of the autophagosome.
Chondrocytes are involved in maintaining a balance
between synthesis and degradation of the ECM. Therefore, cell death
is associated with the breakdown of cartilage (31–33).
In 2004, the term ‘chondroptosis’ was the first to define this type
cell death, which includes classical apoptosis and autophagy
(34). In a number of instances,
the cell switches between the above two responses in a mutually
exclusive manner (35). As for the
non-apoptotic mechanism, numerous studies have examined the roles
of autophagy in normal cartilage metabolism and pathological
conditions. Normal chondrocytes have a certain degree of autophagy
and the aging-associated loss is linked with cell death (12). By contrast, the decreased autophagy
with age may be an explanation for OA and activation of autophagy
may reduce the severity of experimental OA (36). Therefore, in the early stages of
OA, autophagy may be an adaptive response to avoid chondrocyte
death (37). However, in deep
zones of OA cartilage, due to the abnormal subchondral bone
ossification, apoptosis is the only death process observed
(37). In the present study, an
increase in autophagic activity induced by short-term GC treatment
was detected. The results show that only 6 h of chondrocyte
exposure to Dex caused an upregulation in cell autophagy. This
trend reached a maximum following 24 h of treatment. Continuous
exposure to Dex led to the downregulation of autophagic activity.
These resuls were obtained by MDC staining, which identified these
trends in the autophagic activity of cells. The above results
suggest that short-term treatment with GCs may also activate
autophagy, whereas long-term exposure to GCs increases apoptosis.
Autophagy is likely to be a self-protective process in chondrocytes
in response to stimulation with GC.
To further determine the role of pharmacologically
enhanced autophagy associated with the effects of Dex on
chondrocyte viability, Rapa was employed as an autophagy inducer.
The results suggested that, compared with Dex only treatment, Rapa
enhances the viability of the cells after 72 h. Next, western blot
analysis confirmed that the pathological apoptosis markers
caspase-9 and PARP were downregulated by Rapa, while the autophagy
marker LC3 was upregulated. Rapa is a specific inhibitor of mTOR
kinase, which regulates autophagy activation (18). mTOR is a nutrient-sensing kinase
that integrates input from the energy status of the cell.
Therefore, this kinase controls the ribosomal biogenesis and
protein synthesis (16,38). It has been demonstrated that this
kinase is also involved in the cell death of chondrocytes in OA.
Firstly, the activation of mTOR may suppress chondrocyte
proliferation and differentiation by phosphorylation of the
ribosomal protein S6 (39).
Secondly, the inhibition of autophagy by mTOR caused OA-like
changes in gene expression (40).
Furthermore, mTOR inhibition may suppress the expression of A
disintegrin and metalloproteinase with thrombospondin motifs 5 and
interleukin (IL)-1β (36).
Induction of proteinase expression in chondrocytes by IL-1β was
reactive oxygen species (ROS)-dependent. Furthermore, increased ROS
activity has been suggested to inhibit the growth factor expression
and enhance the production of matrix metalloproteinases (41). However, the activation of autophagy
by Rapa markedly reduces the ROS levels induced by IL-1β and
reduces the severity of experimental OA (35,36,42,43).
GCs function as an anti-inflammatory agent. Therefore, in the
present study, it was hypothesized that the apoptosis of
chondrocytes may not be associated with the toxicity of
inflammatory factors, but with the metabolic stress caused by GCs.
Thus, pharmacologically enhanced autophagy by Rapa may act as an
adaptive response to protect the chondrocytes from a relatively
long-term exposure to GCs.
In conclusion, in regard to chondrocytes, the
self-activation of autophagy is a protective mechanism against
apoptosis under short-term treatment with GCs. However, persistent
exposure to GCs may cause the cell to become dysfunctional,
resulting in apoptosis. Furthermore, pharmacologically enhanced
autophagy by Rapa provided novel insights into the treatment of the
adverse effects of GCs on cartilage. Further study is required to
elucidate the interaction between ECM synthesis and autophagy in
chondrocytes.
Acknowledgements
The authors would like to thank Mr Wang Chunlei for
technical assistance.
References
|
1
|
Felson DT and Zhang Y: An update on the
epidemiology of knee and hip osteoarthritis with a view to
prevention. Arthritis Rheum. 41:1343–1355. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Larsson E, Erlandsson Harris H, Larsson A,
Månsson B, Saxne T and Klareskog L: Corticosteroid treatment of
experimental arthritis retards cartilage destruction as determined
by histology and serum COMP. Rheumatology (Oxford). 43:428–434.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hollander JL: Intra-articular
hydrocortisone in arthritis and allied conditions; a summary of two
years’ clinical experience. J Bone Joint Surg Am. 35-A:983–990.
1953.PubMed/NCBI
|
|
4
|
Flanagan J, Casale FF, Thomas TL and Desai
KB: Intra-articular injection for pain relief in patients awaiting
hip replacement. Ann R Coll Surg Engl. 70:156–157. 1988.PubMed/NCBI
|
|
5
|
Friedman DM and Moore ME: The efficacy of
intraarticular steroids in osteoarthritis: a double-blind study. J
Rheumatol. 7:850–856. 1980.PubMed/NCBI
|
|
6
|
Lambert RG, Hutchings EJ, Grace MG,
Jhangri GS, Conner-Spady B and Maksymowych WP: Steroid injection
for osteoarthritis of the hip: a randomized, double-blind,
placebo-controlled trial. Arthritis Rheum. 56:2278–2287. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hainque B, Dominice J, Jaffray P, Ronot X
and Adolphe M: Effects of dexamethasone on the growth of cultured
rabbit articular chondrocytes: relation with the nuclear
glucocorticoid- recepter complex. Ann Rheum Dis. 46:146–152. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Héraud F, Héraud A and Harmand MF:
Apoptosis in normal and osteoarthritic human articular cartilage.
Ann Rheum Dis. 59:959–965. 2000.
|
|
9
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martinet W, Agostinis P, Vanhoecke B,
Dewaele M and De Meyer GR: Autophagy in disease: a double-edged
sword with therapeutic potential. Clin Sci (Lond). 116:697–712.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Caramés B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010.PubMed/NCBI
|
|
13
|
Caramés B, Kiosses WB, Akasaki Y, Brinson
DC, Eap W, Koziol J and Lotz M: Glucosamine activates autophagy in
vitro and in vivo. Arthritis Rheum. 65:1843–1852. 2013.
|
|
14
|
Shigemitsu K, Tsujishita Y, Hara K,
Nanahoshi M, Avruch J and Yonezawa K: Regulation of translational
effectors by amino acid and mammalian target of rapamycin signaling
pathways. Possible involvement of autophagy in cultured hepatoma
cells. J Biol Chem. 274:1058–1065. 1999. View Article : Google Scholar
|
|
15
|
Cardenas ME, Cutler NS, Lorenz MC, Di Como
CJ and Heitman J: The TOR signaling cascade regulates gene
expression in response to nutrients. Genes Dev. 13:3271–3279. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dann SG, Selvaraj A and Thomas G: mTOR
Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and
cancer. Trends Mol Med. 13:252–259. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harrison DE, Strong R, Sharp ZD, Nelson
JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter
CS, Pahor M, Javors MA, Fernandez E and Miller RA: Rapamycin fed
late in life extends lifespan in genetically heterogeneous mice.
Nature. 460:392–395. 2009.PubMed/NCBI
|
|
18
|
Pan T, Rawal P, Wu Y, Xie W, Jankovic J
and Le W: Rapamycin protects against rotenone-induced apoptosis
through autophagy induction. Neuroscience. 164:541–551. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Spilman P, Podlutskaya N, Hart MJ, Debnath
J, Gorostiza O, Bredesen D, Richardson A, Strong R and Galvan V:
Inhibition of mTOR by rapamycin abolishes cognitive deficits and
reduces amyloid-beta levels in a mouse model of Alzheimer’s
disease. PLoS One. 5:e99792010.PubMed/NCBI
|
|
20
|
Song YW, Zhang T and Wang WB:
Gluococorticoid could influence extracellular matrix synthesis
through Sox9 via p38 MAPK pathway. Rheumatol Int. 32:3669–3673.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Buss LW, Anderson C, Westerman E,
Kritzberger C, Poudyal M, Moreno MA and Lakkis FG: Allorecognition
triggers autophagy and subsequent necrosis in the cnidarian
Hydractinia symbiolongicarpus. PLoS One. 7:e489142012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dobson F, Hinman RS, Roos EM, Abbott JH,
Stratford P, Davis AM, Buchbinder R, Snyder-Mackler L, Henrotin Y,
Thumboo J, Hansen P and Bennell KL: OARSI recommended
performance-based tests to assess physical function in people
diagnosed with hip or knee osteoarthritis. Osteoarthritis
Cartilage. 21:1042–1052. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Van Manen MD, Nace J and Mont MA:
Management of primary knee osteoarthritis and indications for total
knee arthroplasty for general practitioners. J Am Osteopath Assoc.
112:709–715. 2012.PubMed/NCBI
|
|
24
|
Schumacher HR and Chen LX: Injectable
corticosteroids in treatment of arthritis of the knee. Am J Med.
118:1208–1214. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bjordal JM, Klovning A, Ljunggren AE and
Slørdal L: Short-term efficacy of pharmacotherapeutic interventions
in osteoarthritic knee pain: A meta-analysis of randomised
placebo-controlled trials. Eur J Pain. 11:125–138. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Olney RC: Mechanisms of impaired growth:
effect of steroids on bone and cartilage. Horm Res. 72(Suppl 1):
30–35. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fubini SL, Todhunter RJ, Burton-Wurster N,
Vernier-Singer M and MacLeod JN: Corticosteroids alter the
differentiated phenotype of articular chondrocytes. J Orthop Res.
19:688–695. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Farkas B, Kvell K, Czömpöly T, Illés T and
Bárdos T: Increased chondrocyte death after steroid and local
anesthetic combination. Clin Orthop Relat Res. 468:3112–3120. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lires-Deán M, Caramés B, Cillero-Pastor B,
Galdo F, López-Armada MJ and Blanco FJ: Anti-apoptotic effect of
transforming growth factor-beta1 on human articular chondrocytes:
role of protein phosphatase 2A. Osteoarthritis Cartilage.
16:1370–1378. 2008.PubMed/NCBI
|
|
32
|
Blanco FJ, López-Armada MJ and Maneiro E:
Mitochondrial dysfunction in osteoarthritis. Mitochondrion.
4:715–728. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mobasheri A: Role of chondrocyte death and
hypocellularity in ageing human articular cartilage and the
pathogenesis of osteoarthritis. Med Hypotheses. 58:193–197. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Roach HI, Aigner T and Kouri JB:
Chondroptosis: a variant of apoptotic cell death in chondrocytes?
Apoptosis. 9:265–277. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Caramés B, Hasegawa A, Taniguchi N, Miyaki
S, Blanco FJ and Lotz M: Autophagy activation by rapamycin reduces
severity of experimental osteoarthritis. Ann Rheum Dis. 71:575–581.
2012.PubMed/NCBI
|
|
36
|
Sasaki H, Takayama K, Matsushita T, Ishida
K, Kubo S, Matsumoto T, Fujita N, Oka S, Kurosaka M and Kuroda R:
Autophagy modulates osteoarthritis-related gene expression in human
chondrocytes. Arthritis Rheum. 64:1920–1928. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Almonte-Becerril M, Navarro-Garcia F,
Gonzalez-Robles A, Vega-Lopez MA, Lavalle C and Kouri JB: Cell
death of chondrocytes is a combination between apoptosis and
autophagy during the pathogenesis of Osteoarthritis within an
experimental model. Apoptosis. 15:631–638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kimball SR and Jefferson LS: Molecular
mechanisms through which amino acids mediate signaling through the
mammalian target of rapamycin. Curr Opin Clin Nutr Metab Care.
7:39–44. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim MS, Wu KY, Auyeung V, Chen Q, Gruppuso
PA and Phornphutkul C: Leucine restriction inhibits chondrocyte
proliferation and differentiation through mechanisms both dependent
and independent of mTOR signaling. Am J Physiol Endocrinol Metab.
296:E1374–E1382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen J, Crawford R and Xiao Y: Vertical
inhibition of the PI3K/Akt/mTOR pathway for the treatment of
osteoarthritis. J Cell Biochem. 114:245–249. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bohensky J, Terkhorn SP, Freeman TA, Adams
CS, Garcia JA, Shapiro IM and Srinivas V: Regulation of autophagy
in human and murine cartilage: hypoxia-inducible factor 2
suppresses chondrocyte autophagy. Arthritis Rheum. 60:1406–1415.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lo YY, Conquer JA, Grinstein S and Cruz
TF: Interleukin-1 beta induction of c-fos and collagenase
expression in articular chondrocytes: involvement of reactive
oxygen species. J Cell Biochem. 69:19–29. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Henrotin YE, Bruckner P and Pujol JP: The
role of reactive oxygen species in homeostasis and degradation of
cartilage. Osteoarthritis Cartilage. 11:747–755. 2003. View Article : Google Scholar : PubMed/NCBI
|