Introduction
Acute myocardial infarction (AMI) is a major
contributor to chronic heart disease leading to mortality in humans
(1,2). Transplantation of mesenchymal stem
cells (MSCs) in the infarcted myocardium is considered to be a
promising therapeutic option to repair the infarcted myocardium and
restore the function of the damaged heart through multiple
mechanisms (3–5). However, poor cell viability following
transplantation significantly limits the therapeutic efficiency of
MSCs (6,7). Mounting evidence has demonstrated
that MSCs undergo apoptosis shortly following transplantation,
which significantly reduces their effectiveness in tissue repair
and regeneration (8,9). Thus, identifying the mechanisms
associated with the apoptosis of MSCs and promoting the survival of
transplanted MSCs may be vital for its successful utilization in
cell therapy.
Hydrogen sulfide (H2S), a poisonous gas
used as a chemical reagent, has recently been proposed to be the
third signaling gasotransmitter besides nitric oxide and carbon
monoxide (10). In mammalian
tissues, endogenous H2S from L-cysteine is catalyzed
primarily by two enzymes, cystathionine γ-lyase (CSE) and
cystathionine β-synthase (CBS) (11). Accumulating evidence suggests that
exogenously applied H2S and inhibition of endogenous
H2S production regulate cell apoptosis in various
systems, including the cardiovascular (12,13),
nervous (14,15), respiratory (16,17)
and pancreatic (18) systems. Xie
et al (19) suggested that
H2S preconditioning protects MSCs against hypoxia and
serum deprivation (hypoxia/SD)-induced apoptosis in vitro
and enhances the efficacy of MSC transplantation in a rat model of
AMI. However, the production of endogenous H2S in MSCs
and its regulatory effect on the apoptosis of MSCs are not yet
completely understood.
In the present study, an apoptosis model induced by
hypoxia/SD was established to determine whether H2S
could be endogenously generated by MSCs, and the role of endogenous
H2S in hypoxia/SD-induced apoptosis of MSCs was
investigated.
Materials and methods
Materials
Low glucose Dulbecco’s Modified Eagle’s Medium
(L-DMEM) and fetal bovine serum (FBS) were purchased from Hyclone
(Logan, UT, USA). L-cysteine, pyridoxal-5′-phosphate, propidium
iodide (PI), RNase, DL-propargylglycine (PPG) and amino-oxyacetate
(AOAA) were provided by Sigma-Aldrich (St. Louis, MO, USA). Rabbit
anti-rat β-actin polyclonal antibody and mouse monoclonal anti-CBS
or -CSE antibodies were obtained from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). The Enhanced Chemiluminescence (ECL)
Western Blotting system was acquired from Millipore (Billerica, MA,
USA), Lipofectamine 2000 was purchased from Invitrogen Life
Technologies (Paisley, Scotland, UK) and polybrene was obtained
from Chemicon (Temecula, CA, USA).
Cell culture and the model of hypoxia and
SD
The MSCs were isolated from Sprague-Dawley rats (~4
weeks old, weight 80 g) obtained from the Laboratory Animal Center
of Anhui Medical University (Hefei, Anhui, China) as previously
described (20). All procedures in
the present study were approved by the Animal Care and Use
Committee of the First Affiliated Hospital of Anhui Medical
University (Hefei, China). Briefly, bone marrow was harvested from
the tibia and femur, plated in L-DMEM supplemented with 15%
inactivated FBS and 100 units/ml of penicillin/streptomycin and
incubated at 37°C in a humidified tissue culture incubator
containing 5% CO2 and 95% air. The medium was replaced
24 h later to discard nonadherent hematopoietic cells. The
adherent, spindle-shaped MSCs were expanded and cultured for no
more than two or three passages. Then they were analyzed by flow
cytometry for the expression of surface markers as described
previously (20). Apoptosis was
induced by hypoxia/SD. Cells were washed with serum-free L-DMEM,
placed in serum-free medium and then incubated in a sealed, hypoxic
GENbox jar fitted with a catalyst (Biomérieux, Marcy l’Étoile,
France) to sequester free oxygen for 3, 6, 12 and 24 h. Oxygen
tension in the medium, measured using a blood gas analyzer, was
33.5 mmHg within 0.5 h after being transferred into the hypoxic
chamber and was maintained at approximately 22–24 mmHg throughout
the experimental time.
Plasmid construction, transfection,
production of lentivirus and transduction
PCR was used to amplify the CSE gene (GenBank™
accession number AY032875) from rat liver tissues using the
following primer set: 5′-GTATGGAGGCACC AACAGGT-3′ and
5′-GTTGGGTTTGTGGGTGTTTC-3′. The amplified CSE gene was subcloned
into the pLVX-IRES-ZsGreen vector by in vitro recombination
methods. A pseudo-lentivirus was produced by transient transfection
of 293FT packaging cells (Type Culture Collection of the Chinese
Academy of Sciences, Shanghai, China). One day prior to the
transfection, 1.6×106 293FT cells were plated in 6-cm
dishes. Then cells were cotransfected with either 1.7 μg of the
pLVX-IRES-ZsGreen vector or pLVX-IRES-ZsGreen-CSE, 1.13 μg of pCMV
Δ8.91 and 0.57 μg of pMD.G using Lipofectamine 2000. Culture
supernatants were harvested 72 h after transfection and filtered
through a 0.45 μm low protein binding polysulfonic filter
(Millipore, Bedford, MA, USA). For transduction, 2×106
MSC cells were seeded into 10 cm dishes and incubated with
lentiviruses and 8 μg/ml of polybrene in the incubator for 48 h.
Then the transduction efficiency was detected by the green
fluorescence expression using an inverted microscope (TE2000-U;
Nikon, Tokyo, Japan).
Flow cytometric analysis of
apoptosis
Treated MSCs were digested with trypsin (2.5 g/l)
and centrifuged at 250 × g for 5 min, and then the supernatant was
removed. Cells were washed twice with phosphate-buffered saline
(PBS) and fixed with 70% ethanol at −20°C overnight. Cells were
then centrifuged at 250 × g for 5 min, washed in PBS twice and
adjusted to a concentration of 1×106 cells/ml. A volume
of 0.5 ml of RNase (1 mg/ml in PBS; Sigma-Aldrich) was added into
0.5-ml cell samples and incubated at 37°C for 30 min. Following
mixing with PI (at a final concentration of 50 mg/l), mixed cells
were filtered and incubated in the dark at 4°C for 30 min prior to
flow cytometric analysis (BD FACS Calibur; Beckman Coulter, Miami,
FL, USA). In the DNA histogram, the amplitude of the sub-G1 DNA
peak represents the number of apoptotic cells.
Measurement of H2S in the cell
culture supernatant
The basis of the assay was that the H2S
produced in the incubate reacts with zinc acetate to form zinc
sulfide, which then dissolves in a hydrochloride acid solution of
N, N-dimethyl-p-phenylenediamine sulfate (NNDPD), yielding, in the
presence of ferric chloride, methylene blue, which is quantitated
spectrophotometrically. Cell culture supernatant (310 μl) was mixed
with trichloroacetic acid (20% w/v, 60 μl), zinc acetate (2% w/v,
30 μl), NNDPD (20 mM, 40 μl) in 7.2 M HCl and FeCl3 (30
mM, 30 μl) in 1.2 M of HCl. The absorbance of the resulting
solution (670 nm) was measured 15 min thereafter by
spectrophotometry (DU800; Beckman Coulter). H2S
concentration was calculated against a calibration curve of
NaHS.
Assay of the activity of H2S
synthesizing enzymes
The MSCs were homogenized in 50 mM of ice-cold
potassium phosphate buffer (pH 6.8). The reaction mixture contained
100 mM of potassium phosphate buffer (pH 7.4), L-cysteine (20 μl,
10 mM), pyridoxal 5′-phosphate (20 μl, 2 mM), saline (30 μl) and
11% w/v cell homogenate (430 μl). The reaction was performed in
tightly stoppered cryovial test tubes and initiated by transferring
the tubes from ice to an agitating water bath at 37°C. Following
incubation for 30 min, 1% w/v zinc acetate (250 μl) was added to
trap evolved H2S followed by 10% v/v trichloroacetic
acid (250 μl) to denature the protein and stop the reaction.
Subsequently, NNDPD (20 mM; 133 μl) in 7.2 M of HCl was added,
immediately followed by FeCl3 (30 mM; 133 μl) in 1.2 M
HCl. The absorbance of the resulting solution (670 nm) was measured
by spectrophotometry. The H2S concentration was
calculated against a calibration curve of NaHS, and H2S
synthesizing activity is expressed as nanomoles of H2S
formed per gram of protein (determined using the Bradford assay)
per min (nmol/min/g protein).
Protein extraction and western blot
analysis
For western blot analysis, cell lysates were
prepared in the lysis buffer. Following centrifugation at 15,000 ×
g for 10 min, the supernatant was analyzed by western blotting.
Total cell protein concentration was determined using the DC
Protein Assay kit (Bio-Rad, Hercules, CA, USA). SDS-polyacrylamide
gel electrophoresis was performed on 5% stacking and 12% resolving
gel with low-range molecular weight standards (Solarbio, Beijing,
China). Equal amounts of protein were loaded in each lane with
loading buffer (Beyotime Biotechnology, Haimen, Jiangsu, China)
containing 0.1 M Tris (pH 6.8), 20% glycerol, 10% mercaptoethanol,
4% SDS and 0.2% bromophenol blue. Following electrophoresis, the
proteins were transferred to an Immobilon-NC membrane (Millipore,
Billerica, MA, USA). Following this, the membranes were blocked
with Tris-buffered saline with Tween-20 (TBST; 50 mM of Tris-HCl,
pH 7.4, 0.15 M of NaCl, 0.1% Tween-20) containing 5% BSA
(Sigma-Aldrich) for 2 h. Following this, the membranes were
incubated with primary antibodies at 4°C overnight. Following
washing with TBST, the membranes were incubated with anti-rabbit
IgG, HRP-linked antibody (CST; 1:1,000) at room temperature for 2
h. The membranes were washed again and developed with an ECL system
(Millipore) followed by apposition of the membranes with
autoradiographic films (Kodak, Shanghai, China). The optical
density of the protein band on western blots was calculated by
Quantity One software (Bio-Rad).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Differences among groups were tested by one-way analysis
of variance. Comparisons between two groups were evaluated using
post-hoc tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
Hypoxia/SD induces cell apoptosis and
inhibits endogenous H2S production in MSCs
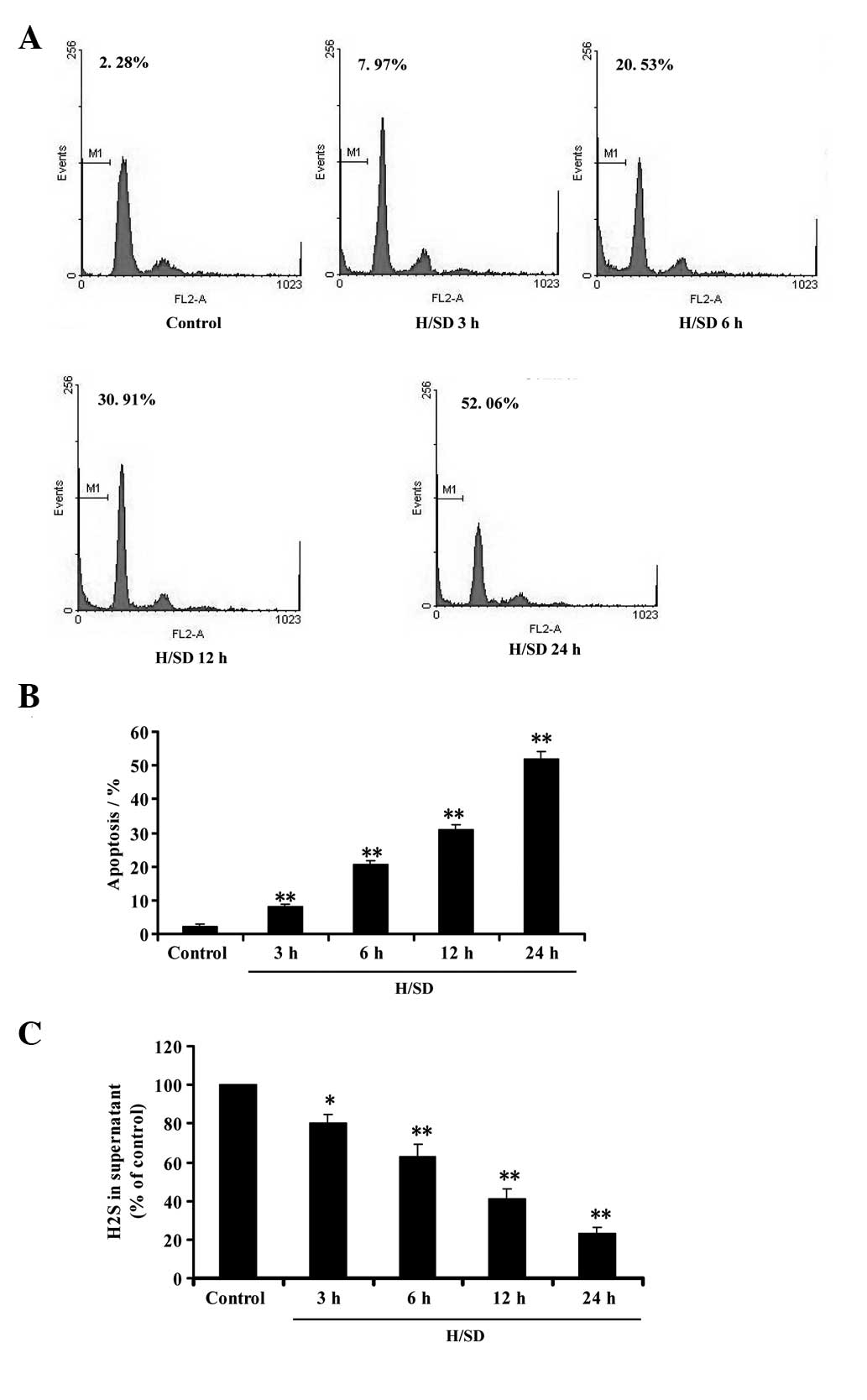
Flow cytometric analysis was used to investigate the
effect of hypoxia/SD on apoptosis in MSCs. As shown in Fig. 1A and B, the treatment of MSCs with
hypoxia/SD (3–24 h) significantly induced the apoptosis of MSCs in
a time-dependent manner. At the same time, we evaluated whether
hypoxia/SD affects the generation of endogenous H2S in
MSCs. Following exposure of MSCs to hypoxia/SD (3–24 h), the
content of H2S in culture supernatant was
time-dependently decreased (Fig.
1C), indicating that hypoxia/SD was able to inhibit endogenous
H2S production in MSCs. These data imply that
hypoxia/SD-induced apoptosis may be associated with the inhibition
of endogenous H2S generation in MSCs.
H2S is produced by CSE in
MSCs
CBS and CSE are the two key enzymes involved in
H2S formation from L-cysteine. Western blot analysis was
used to investigate whether CBS and/or CSE are present in MSCs. As
shown in Fig. 2A, CSE was detected
in MSCs by western blot analysis; however, western blot analysis
did not reveal the expression of CBS. This result indicated that
CSE, but not CBS, is expressed in MSCs.
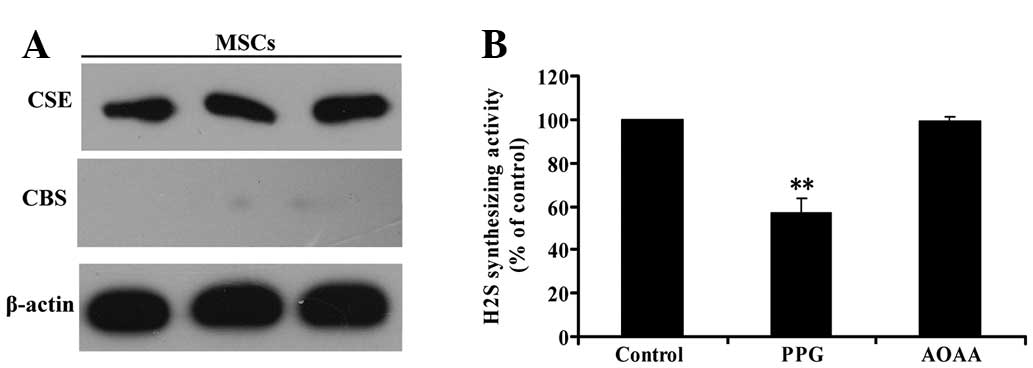 | Figure 2CSE, but not CBS, is involved in
H2S generation in MSCs. (A) Three samples of protein
from MSCs were extracted and analyzed for CSE and CBS expression by
western blot analysis (β-actin was used as an internal control).
(B) MSCs were homogenized and, respectively, pretreated with AOAA
(300 μmol/l), the CSB inhibitor or PPG (5 mmol/l), the CSE
inhibitors, for 30 min prior to adding the reaction mixture of
H2S generation. Next, H2S synthesizing
activity from added L-cysteine in MSC homogenate was evaluated.
Values are the mean ± SEM of three independent experiments.
**P<0.01, compared with the control group. CSE,
cystathionine γ-lyase; CBS, cystathionine β-synthase;
H2S, hydrogen sulfide; MSCs, mesenchymal stem cells;
AOAA, amino-oxyacetate; PPG, DL-propargylglycine. |
To confirm that CSE is the major enzyme responsible
for endogenous H2S generation in MSCs, we explored the
effects of the inhibitor of CBS, AOAA, and the inhibitor of CSE,
PPG, on H2S synthesis in MSCs. H2S
synthesizing activity in MSCs was evaluated by the production of
H2S from added L-cysteine in the lysates of MSCs. The
activity of H2S synthesis in MSCs was significantly
inhibited by pretreatment with PPG (Fig. 2B) for 30 min prior to the
application of L-cysteine. By contrast, pretreatment with AOAA had
no effect on the level of H2S production (Fig. 2B), indicating that CSE, but not
CBS, is involved in H2S generation in MSCs.
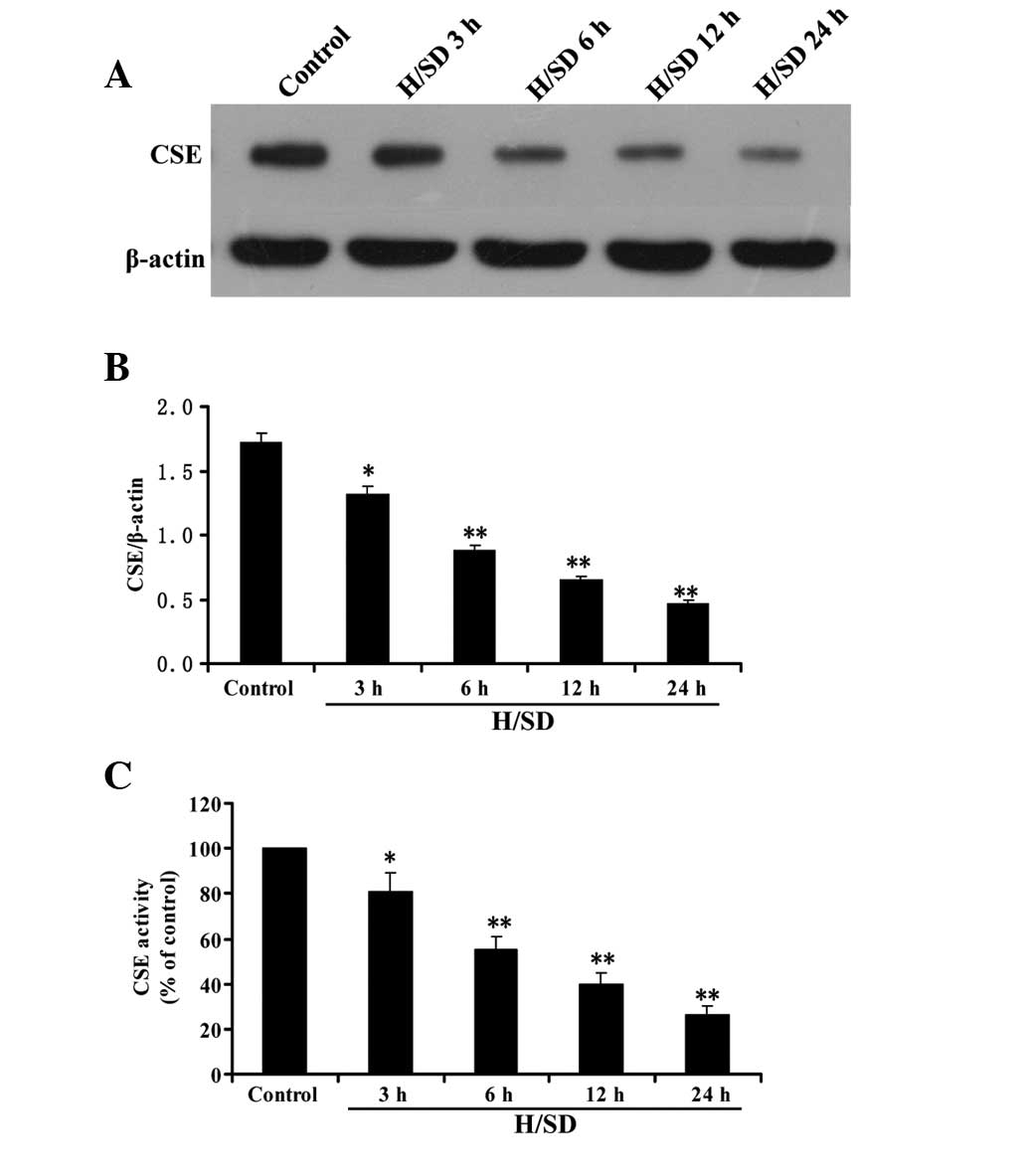
Hypoxia/SD inhibits CSE expression and
activity in MSCs
As shown in Fig. 3A and
B, following treatment of MSCs with hypoxia/SD (3–24 h), CSE
expression decreased in a time-dependent manner. Consistent with
the results of CSE expression, the activity of CSE in MSCs was
decreased by treatment with hypoxia/SD (3–24 h) in a time-dependent
manner (Fig. 3C). These data
indicate that inhibition of CSE expression and activity in MSCs
contributes to the hypoxia/SD-elicited decrease in endogenous
H2S production.
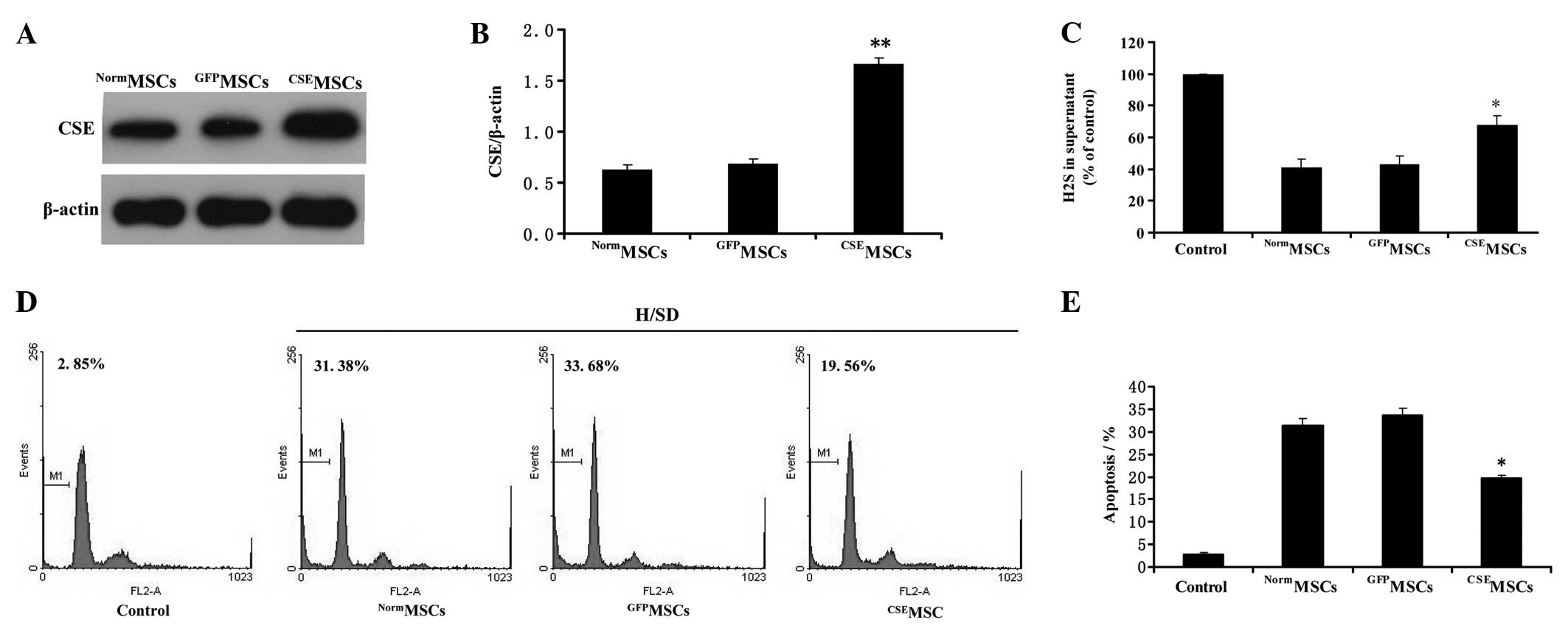
CSE overexpression protects MSCs from
hypoxia/SD-induced apoptosis in MSCs
To further explore the regulatory role of the
endogenous CSE/H2S system in hypoxia/SD-induced
apoptosis in MSCs, we evaluated the effects of CSE overexpression
on hypoxia/SD-induced decreases in endogenous H2S
generation and apoptosis in MSCs. CSE overexpression was mediated
by lentiviral transduction in MSCs. Western blot analysis
demonstrated that CSE expression in the MSCs infected by the
pLV-ZsGreen-CSE lentivirus (CSEMSCs) was upregulated by
more than 2.4-fold compared with the MSCs infected by the
pLV-ZsGreen lentivirus (GFPMSCs) or untransduced MSCs
(NormMSCs) (Fig. 4A and
B). Next, the modified MSCs were treated under hypoxia/SD for
12 h. As shown in Fig. 4C, the
content of H2S in culture supernatant was significantly
increased in the CSEMSC group compared with the
NormMSCs or GFPMSCs under hypoxia/SD for 12
h. Furthermore, we demonstrated that CSEMSCs had a
significant lower proportion of apoptosis (Fig. 4D and E) compared with
NormMSCs or GFPMSCs groups under hypoxia/SD
for 12 h. Overall, these data indicate that the upregulation of the
CSE/H2S system protects MSCs from hypoxia/SD-induced
apoptosis in MSCs.
CSE inhibitor deteriorates
hypoxia/SD-induced apoptosis in MSCs
To further confirm whether hypoxia/SD-induced
apoptosis occurs via the endogenous CSE/H2S system in
MSCs, we inhibited H2S production by applying the CSE
inhibitor PPG in the presence of hypoxia/SD for 12 h. As shown in
Fig. 5A, PPG (5 mmol/l) not only
reduced H2S generation but also exacerbated the
inhibition of H2S generation elicited by hypoxia/SD for
12 h. Furthermore, cell apoptosis (Fig. 5B and C) under hypoxia/SD was also
significantly deteriorated by pretreating MSCs with the CSE
inhibitor PPG (5 mmol/l) for 30 min. The CSE inhibitor PPG (5
mmol/l) treatment alone, however, had no effect on the apoptosis of
MSCs (Fig. 5B and C). Overall,
these data indicate that inhibition of the CSE/H2S
system deteriorates hypoxia/SD-induced apoptosis in MSCs.
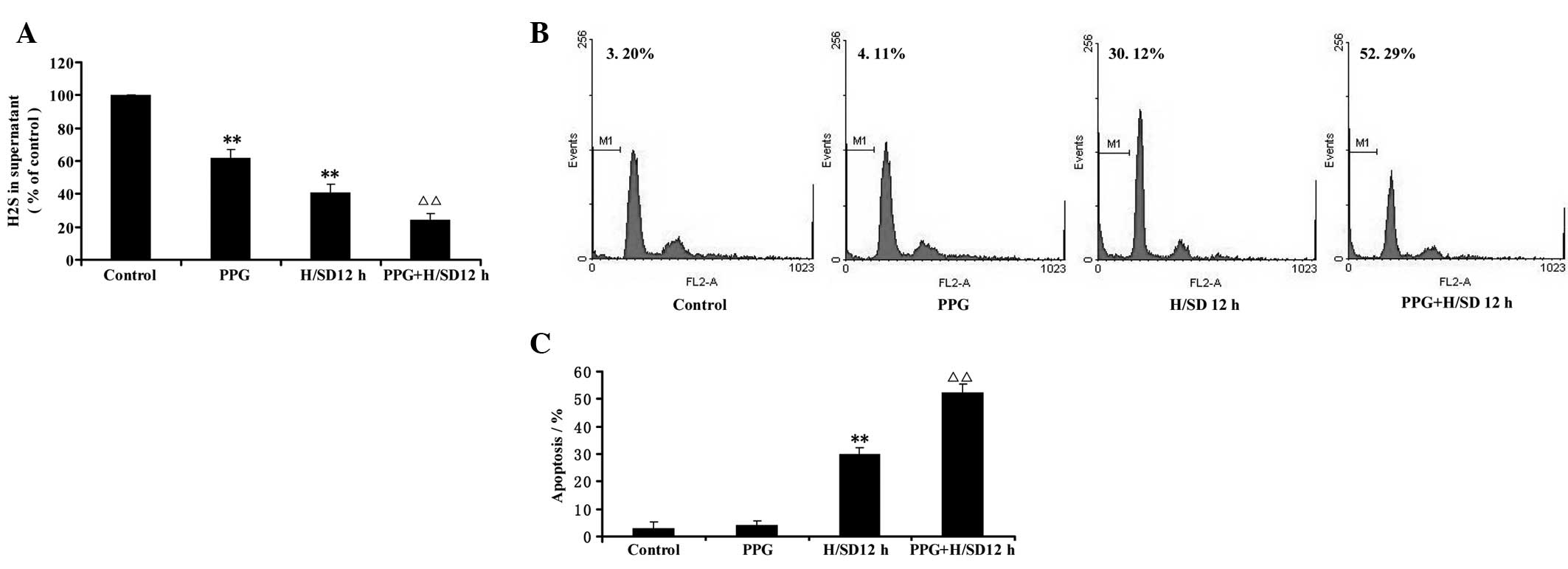 | Figure 5Effect of PPG, a blocker of CSE
activity, on hypoxia/SD-induced decreases in endogenous
H2S generation and apoptosis in MSCs. (A) MSCs were
pretreated with the CSE inhibitor PPG (5 mmol/l) for 30 min prior
to treatment with hypoxia/SD for 12 h and the content of
H2S in the culture supernatant was measured by the
N,N-dimethyl-p-phenylenediamine sulfate method as described in
Materials and methods. (B) Cell apoptosis was determined by flow
cytometric analysis. (C) Quantitative analysis of the rate of
apoptosis. Values are the mean ± SEM of three independent
experiments. **P<0.01, versus control group;
ΔΔP<0.01, versus treatment with hypoxia/SD for 12 h
alone group. PPG, DL-propargylglycine; CSE, cystathionine γ-lyase;
SD, serum deprivation; H2S, hydrogen sulfide; MSCs,
mesenchymal stem cells; H/SD, hypoxia/serum deprivation. |
Discussion
The present study demonstrated that CSE, but not
CBS, was expressed in MSCs and involved in the endogenous
generation of H2S in MSCs. The treatment of MSCs with
hypoxia/SD led to: i) time-dependent apoptosis in MSCs, ii) a
decrease in endogenous H2S generation, and iii)
inhibition of CSE expression and activity. Furthermore, we also
demonstrated that overexpression of CSE not only markedly prevented
hypoxia/SD-induced decreases of endogenous H2S
production but also protected MSCs from apoptosis, while inhibition
of CSE by its potent inhibitors significantly deteriorated the
effect of hypoxia/SD in MSCs. Collectively, these findings suggest
that hypoxia/SD induces apoptosis in MSCs by decreasing endogenous
H2S production via inhibiting the endogenous
CSE/H2S system.
Endogenous H2S is now regarded as a novel
signaling gasotransmitter and is important physiologically and
physiopathologically in vivo and in vitro (21–24).
MSCs, separated from bone marrow, periosteum, cord blood, skeletal
muscle and adipose tissue, are capable of self-renewal (25)and multiple paths of differentiation
(26) and have been demonstrated
as an ideal cell source for the treatment of AMI (3–5).
Recent data demonstrated that H2S was able to protect
MSCs against hypoxia/SD-induced apoptosis in vitro and
enhances the efficacy of MSC transplantation in a rat model of AMI
(19). In the present study, we
demonstrated that MSCs were able to generate H2S and
that this gaseous mediator is important in regulating
hypoxia/SD-induced apoptosis. Two pyridoxal-5′-phosphate-dependent
enzymes, CBS and CSE, are responsible for the majority of the
endogenous production of H2S in mammalian tissues
(11). CBS is mainly expressed in
the nervous system, whereas CSE appears to be predominant in the
cardiovascular system (27). We
demonstrated that CSE, but not CBS, was detected in MSCs and that
conversion of L-cysteine to H2S in the lysates of MSCs
was inhibited by PPG, the inhibitor of CSE. These data suggest that
CSE is the main enzyme involved in the generation of H2S
in MSCs. Recently, Shibuya et al (28) demonstrated that endogenous
H2S was also able to be produced by 3-mercaptopyruvate
sulfurtransferase (3-MST) along with cysteine aminotransferase in
the brain. Further study needs to be carried out in the future to
address whether MSCs are able to generate H2S by
catalyzing 3-MST.
Despite their several advantages for the treatment
of AMI, a major challenge of MSC therapy is that transplanted cells
undergo apoptosis (8,9) as they are exposed to an extremely
harsh microenvironment in the infarcted heart. The efficiency of
MSC transplantation is limited by the low viability of MSCs
(29). Therefore, elucidating the
molecular mechanisms underlying the apoptosis of MSCs may lead to
important insights into the pathogenesis and treatment of AMI.
H2S deficiency was observed in atherosclerosis (30), ischemia-reperfusion injury
(31), hypertension (32), gastric mucosal injury and liver
cirrhosis (33). However, to the
best of our knowledge, there is no information concerning
H2S generation in MSCs under hypoxia/SD conditions. In
the present study, we demonstrated that the exposure of MSCs to
hypoxia/SD led to a significant decrease in H2S
generation. We further demonstrated that hypoxia/SD inhibited the
expression and activity of CSE in MSCs. These data indicate that
hypoxia/SD reduces H2S generation by inhibiting the
expression and activity of CSE.
In the present study, we demonstrated that the
treatment of MSCs with hypoxia/SD induced marked cell apoptosis in
a time-dependent manner, which was consistent with the previous
study (34). Taking into account
the fact that hypoxia/SD decreases endogenous H2S
generation and inhibits CSE expression and activity in MSCs, we
hypothesized that hypoxia/SD-induced MSCs apoptosis was associated
with decreased endogenous H2S production. To elucidate
the contribution of decreases in endogenous H2S to
hypoxia/SD-induced MSC apoptosis, we investigated the effect of CSE
overexpression and CSE inhibition on hypoxia/SD-induced apoptosis
in MSCs. Our results demonstrated that overexpression of CSE not
only markedly prevented hypoxia/SD-induced decreases in endogenous
H2S production but also protected MSCs from apoptosis in
MSCs. Together with the aforementioned results that H2S
preconditioning protects MSCs against hypoxia and SD-induced
apoptosis in vitro, we suggest that downregulating the
endogenous CSE/H2S system contributes to
hypoxia/SD-induced MSC apoptosis. Furthermore, pretreatment with
PPG, the inhibitor of CSE, caused a significant reduction in
H2S generation and deteriorated hypoxia/SD-induced
apoptosis in MSCs, further confirming the contribution of decreases
in endogenous H2S production to hypoxia/SD-induced
apoptosis in MSCs.
In summary, we have demonstrated that hypoxia/SD
inhibits the expression and activity of CSE, a key enzyme for
H2S synthesis in MSCs. Overexpression of CSE is able to
markedly prevent hypoxia/SD-induced decreases in endogenous
H2S generation and protect MSCs from apoptosis. Whereas,
inhibition of CSE deteriorates the effect of hypoxia/SD in MSCs.
These findings suggest that hypoxia/SD-induced apoptosis in MSCs
occurs via the inhibition of the endogenous CSE/H2S
system. These data also establish a role for endogenous
H2S in the regulation of hypoxia/SD-induced apoptosis
and identify the CSE/H2S system as a novel therapeutic
target for hypoxia/SD-induced apoptosis in MSCs.
Acknowledgements
The authors would like to thank Dr Houfeng Zheng
from the Jewish General Hospital, McGill University in Canada for
assistance with the preparation of this manuscript.
References
|
1
|
Lloyd-Jones DM, Larson MG, Leip EP, et al:
Lifetime risk for developing congestive heart failure: the
Framingham Heart Study. Circulation. 106:3068–3072. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang H, Wei YJ and Hu SS: Intraoperative
cell transplantation for congestive heart failure: experience in
China. Semin Thorac Cardiovasc Surg. 20:126–130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kovacic JC and Graham RM: Stem-cell
therapy for myocardial diseases. Lancet. 363:1735–1736. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fukuda K and Yuasa S: Stem cells as a
source of regenerative cardiomyocytes. Circ Res. 98:1002–1013.
2006.PubMed/NCBI
|
|
5
|
Nesselmann C, Ma N, Bieback K, et al:
Mesenchymal stem cells and cardiac repair. J Cell Mol Med.
12:1795–1810. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai W, Hale SL, Martin BJ, et al:
Allogeneic mesenchymal stem cell transplantation in postinfarcted
rat myocardium: short- and long-term effects. Circulation.
112:214–223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Müller-Ehmsen J, Krausgrill B, Burst V, et
al: Effective engraftment but poor mid-term persistence of
mononuclear and mesenchymal bone marrow cells in acute and chronic
rat myocardial infarction. J Mol Cell Cardiol. 41:876–884.
2006.PubMed/NCBI
|
|
8
|
Zhu W, Chen J, Cong X, Hu S and Chen X:
Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem
cells. Stem Cells. 24:416–425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robey TE, Saiget MK, Reinecke H and Murry
CE: Systems approaches to preventing transplanted cell death in
cardiac repair. J Mol Cell Cardiol. 45:567–581. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Papapetropoulos A, Pyriochou A, Altaany Z,
et al: Hydrogen sulfide is an endogenous stimulator of
angiogenesis. Proc Natl Acad Sci USA. 106:21972–21977. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sen U, Givvimani S, Abe OA, Lederer ED and
Tyagi SC: Cystathionine beta-synthase and cystathionine gamma-lyase
double gene transfer ameliorate homocysteine-mediated mesangial
inflammation through hydrogen sulfide generation. Am J Physiol Cell
Physiol. 300:C155–C163. 2011. View Article : Google Scholar
|
|
12
|
Lu F, Xing J, Zhang X, et al: Exogenous
hydrogen sulfide prevents cardiomyocyte apoptosis from cardiac
hypertrophy induced by isoproterenol. Mol Cell Biochem. 381:41–50.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calvert JW, Coetzee WA and Lefer DJ: Novel
insights into hydrogen sulfide--mediated cytoprotection. Antioxid
Redox Signal. 12:1203–1217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kimura H: Hydrogen sulfide: its production
and functions. Exp Physiol. 96:833–835. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang XQ, Ren YK, Zhou CF, et al: Hydrogen
sulfide prevents formaldehyde-induced neurotoxicity to PC12 cells
by attenuation of mitochondrial dysfunction and pro-apoptotic
potential. Neurochem Int. 61:16–24. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baskar R, Li L and Moore PK: Hydrogen
sulfide-induces DNA damage and changes in apoptotic gene expression
in human lung fibroblast cells. FASEB J. 21:247–255. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen YH, Wang PP, Wang XM, et al:
Involvement of endogenous hydrogen sulfide in cigarette
smoke-induced changes in airway responsiveness and inflammation of
rat lung. Cytokine. 53:334–341. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Taniguchi S, Kang L, Kimura T and Niki I:
Hydrogen sulphide protects mouse pancreatic beta-cells from cell
death induced by oxidative stress, but not by endoplasmic reticulum
stress. Br J Pharmacol. 162:1171–1178. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie X, Sun A, Zhu W, et al:
Transplantation of mesenchymal stem cells preconditioned with
hydrogen sulfide enhances repair of myocardial infarction in rats.
Tohoku J Exp Med. 226:29–36. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang A, Shen F, Liang Y and Wang J:
Marrow-derived MSCs and atorvastatin improve cardiac function in
rat model of AMI. Int J Cardiol. 150:28–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pae HO, Lee YC, Jo EK and Chung HT: Subtle
interplay of endogenous bioactive gases (NO, CO and H(2)S) in
inflammation. Arch Pharm Res. 32:1155–1162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wallace JL: Physiological and
pathophysiological roles of hydrogen sulfide in the
gastrointestinal tract. Antioxid Redox Signal. 12:1125–1133. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kimura Y, Goto Y and Kimura H: Hydrogen
sulfide increases glutathione production and suppresses oxidative
stress in mitochondria. Antioxid Redox Signal. 12:1–13. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mani S, Li H, Untereiner A, et al:
Decreased endogenous production of hydrogen sulfide accelerates
atherosclerosis. Circulation. 127:2523–2534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barry FP and Murphy JM: Mesenchymal stem
cells: clinical applications and biological characterization. Int J
Biochem Cell Biol. 36:568–584. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Short B, Brouard N, Occhiodoro-Scott T,
Ramakrishnan A and Simmons PJ: Mesenchymal stem cells. Arch Med
Res. 34:565–571. 2003. View Article : Google Scholar
|
|
27
|
Tan BH, Wong PT and Bian JS: Hydrogen
sulfide: a novel signaling molecule in the central nervous system.
Neurochem Int. 56:3–10. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shibuya N, Tanaka M, Yoshida M, et al:
3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and
bound sulfane sulfur in the brain. Antioxid Redox Signal.
11:703–714. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang W, Li W, Ou L, et al:
Polyethylenimine-mediated gene delivery into human bone marrow
mesenchymal stem cells from patients. J Cell Mol Med. 15:1989–1998.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mani S, Untereiner A, Wu L and Wang R:
Hydrogen Sulfide and the Pathogenesis of Atherosclerosis. Antioxid
Redox Signal. May 21–2013.(Epub ahead of print).
|
|
31
|
Nicholson CK and Calvert JW: Hydrogen
sulfide and ischemia-reperfusion injury. Pharmacol Res. 62:289–297.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang G, Wu L, Jiang B, et al: H2S as a
physiologic vasorelaxant: hypertension in mice with deletion of
cystathionine gamma-lyase. Science. 322:587–590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Łowicka E and Bełtowski J: Hydrogen
sulfide (H2S)-the third gas of interest for pharmacologists.
Pharmacol Rep. 59:4–24. 2007.PubMed/NCBI
|
|
34
|
Nie Y, Han BM, Liu XB, et al:
Identification of MicroRNAs involved in hypoxia- and serum
deprivation-induced apoptosis in mesenchymal stem cells. Int J Biol
Sci. 7:762–768. 2011. View Article : Google Scholar : PubMed/NCBI
|