Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory
autoimmune disease, associated with focal and systemic bone loss.
Structural changes such as joint damage and osteoporosis are the
most suitable symptoms for distinguishing RA from other diseases
(1). The level of tumor necrosis
factor-α (TNF-α) is markedly increased in most patients with RA,
and thus, the protein is considered a main pathological player in
inflammation. As a result, many TNF-α inhibitors, including
infliximab (Remicade®), adalimumab (Humira®),
and etanercept (Enbrel®), were developed and have been
widely used to treat RA (2,3).
Etanercept, one of the most commonly used TNF-α
inhibitors, consists of two human TNF receptor 2 (TNFR2)
extracellular domains conjugated to the Fc portion of the human
IgG1. The anti-inflammatory effect of etanercept in patients with
RA is widely accepted (4–7). Although a previous study indicated
that inhibiting inflammation in RA is insufficient to inhibit bone
destruction (8), another study
showed that TNF-α blockers directly mitigate osteoporosis (9). It was also suggested that TNF-α
inhibitors prevent resorption of the bone adjacent to the joints
(10).
Despite the efficiency of TNF-α inhibition in RA
treatment, it is commonly not feasible to prescribe TNF-α
inhibitors to patients with early RA, due to its high cost and
insurance regulations (3,11). British guidelines require
limitation of prescriptions of TNF-α inhibitors to patients with
active RA who do not have a satisfactory response to at least two
disease-modifying antirheumatic drugs (DMARDs) (12). However, since it has been reported
that etanercept is effective when administered to patients with
early RA (5,13), the importance of using this drug in
the treatment of early RA is being reassessed (14). Furthermore, there has been limited
clinical research on the anti-osteoporotic effects of TNF-α
inhibitors in patients with early RA. In this study, we simulated
this clinical condition in mice by transiently injecting etanercept
at the early stage of CIA induction.
Since TNF inhibitors have become popular drugs for
the treatment of RA, clinicians are increasingly interested in the
subgroup of the patients who show a refractory response to
etanercept. One explanation for refractoriness is the development
of anti-drug antibody (ADA) against the protein, as part of the
body’s physiological reaction. Not only the mouse chimeric form of
the monoclonal antibody, infliximab, but also humanized drugs such
as adalimumab, are reported to induce the development of ADA.
However, etanercept is less known to induce ADA and less studied in
comparison to infliximab and adalimumab.
In this study, we established a mouse arthritic
model in which ADA production was investigated at various doses of
etanercept challenge. Furthermore, we investigated whether mice
with ADA show reduced focal and systemic osteoporosis as well as
inflammation, and explored the underlying mechanisms. In addition,
we studied the effect of etanercept in early RA by injecting CIA
mice with etanercept during the early stages of RA.
Materials and methods
Induction of CIA and assessment of
severity
All procedures on animals were in accordance with
the Laboratory Animals Welfare Act, the Guide for the Care and Use
of Laboratory Animals, and the Guidelines and Policies for Rodent
Experiments provided by the Institutional Animal Care and Use
Committee (IACUC) in the School of Medicine, The Catholic
University of Korea [Catholic University Medical College
(CUMC-2011-0010-03)]. Male DBA1/J mice (6 weeks old; Orient Bio
Inc, Seongnam, Korea) were immunized by intradermal injection into
the base of the tail of bovine type II collagen (100 μg/mouse;
Chondrex, Inc., Redmond, WA, USA), emulsified in Freund’s complete
adjuvant (Arthrogen-CIA®, Chondrex, Inc). CIA symptoms
were evident between 17 and 21 days following the first
immunization. The incidence and severity of CIA were monitored and
scored as previously described (15). The CIA incidence was expressed as
the percentage of swollen paw in the 4 paws of each mouse.
Etanercept treatment
Mice with CIA were given an intraperitoneal
injection of either phosphate-buffered saline (PBS) or 25, 100 and
400 μg etanercept (Enbrel®; Pfizer, New York, NY, USA) 3
times/week, from day 7 to day 28 after the first immunization. No
additional treatment was provided until sacrifice, performed at
week 10 after the first immunization.
Histological evaluation of arthritis
The hind leg of each mouse was isolated and fixed in
10% formalin. After decalcification in hydrochloric acid, samples
were embedded in paraffin. The sections were stained with
hematoxylin and eosin (H&E), safranin O, and toluidine blue.
The degree of inflammation was determined based on H&E staining
using the following scoring scheme: 0, no inflammation; 1, mild
thickening of the lining layer or some infiltrating cells in the
sublining layer; 2, mild thickening of the lining layer and some
infiltrating cells in the sublining layer; 3, thickening of the
lining layer, influx of cells in the sublining layer, and existence
of cells in the synovial space; and 4, synovium highly infiltrated
with numerous inflammatory cells. Cartilage damage was scored based
on safranin O and toluidine blue staining according to the
following scheme: 0, no destruction; 1, minimal erosion, limited to
single spots; 2, slight to moderate erosion in a limited area; 3,
more extensive erosion; and 4, general destruction.
Tartrate-resistant acid phosphatase (TRAP) staining was performed
using a commercial kit (Sigma-Aldrich, St. Louis, MO, USA) as
described by the manufacturer, except for hematoxylin
counterstaining. TRAP+ multinucleated cells with ≥3
nuclei were counted as osteoclasts. All histological assessments
were performed by 2 independent blinded observers.
Quantification of cytokine levels
At 2 and 3 weeks following the first collagen
immunization, ~100 μl of venous blood were taken from the orbital
sinus of anesthetized mice. Following incubation at room
temperature for 1 h, blood samples were centrifuged for 20 min at
15,000 g. Serum was transferred into new tubes and stored at −80°C.
The sera were analyzed with a Milliplex® MAP Mouse
Cytokine/Chemokine kit that uses a Luminex xMAP detection system
(Millipore, Bedford, MA, USA). Quantification of data was performed
using the Masterplex QT version 4.0 software (MiraiBio Inc., Tokyo,
Japan).
Microfocal computed tomography (micro-CT)
analyses
Micro-CT analyses of the distal femoral and proximal
tibial metaphyses were performed with a desktop microcomputer
tomography scanner (SkyScan 1172; Bruker-microCT, Kontich,
Belgium). The samples were fixed in 3.7% formaldehyde for >24 h
and were scanned through a 0.5-mm-thick filter using a 141 μA
current, 70 kV source voltage, and an exposure time of 590 msec. To
consistently set the trabecular bone region range, data from each
sample were resampled with the CTAn application following
reconstruction of the scanned images with the NRecon application
(both from BRUKER-MICROCT). The morphometric parameters percentage
of bone volume, trabecular thickness, trabecular number, trabecular
separation and bone surface density were measured by the CTAn
application. Bone mineral density was measured in 77 continuous
slices.
Measurement of serum anti-etanercept
antibody levels
Sera were obtained from all mice at sacrifice, and
the concentration of the anti-etanercept antibody was measured by
enzyme-linked immunosorbent assay (ELISA) as previously described
(16), with minor modifications.
Briefly, 250 ng/50 μl etanercept (0.1 M sodium carbonate, pH 9.6)
were coated on the surface of a microtiter plate overnight at 37°C.
The coated wells were blocked with 2% skim milk in PBS for 1 h at
37°C and rinsed with washing buffer (0.1% Tween-20 in PBS).
Serially diluted serum samples (1:1,000–1:100,000) were added to
the wells for 2 h. The wells were then washed 3 times with washing
buffer. Horseradish peroxidase (HRP)-conjugated anti-mouse IgG at a
1:5,000 dilution in PBS was added and incubated for 1 h. After 5
washes with washing buffer, the tetramethyl benzidine reaction was
initiated, and H2SO4 was used as the stop
solution. The absorbance (optical density; OD) was measured at 450
nm.
Results
Etanercept attenuates arthritis in CIA
mice
Since bone damage starts at the early stage of the
disease, it is advised to inject etanercept in CIA mice at the
early stages to examine its anti-osteoporotic effect. Thus, we
performed 9 intraperitoneal injections of etanercept to CIA mice, 3
times per week for 3 weeks, starting from day 7 after the first
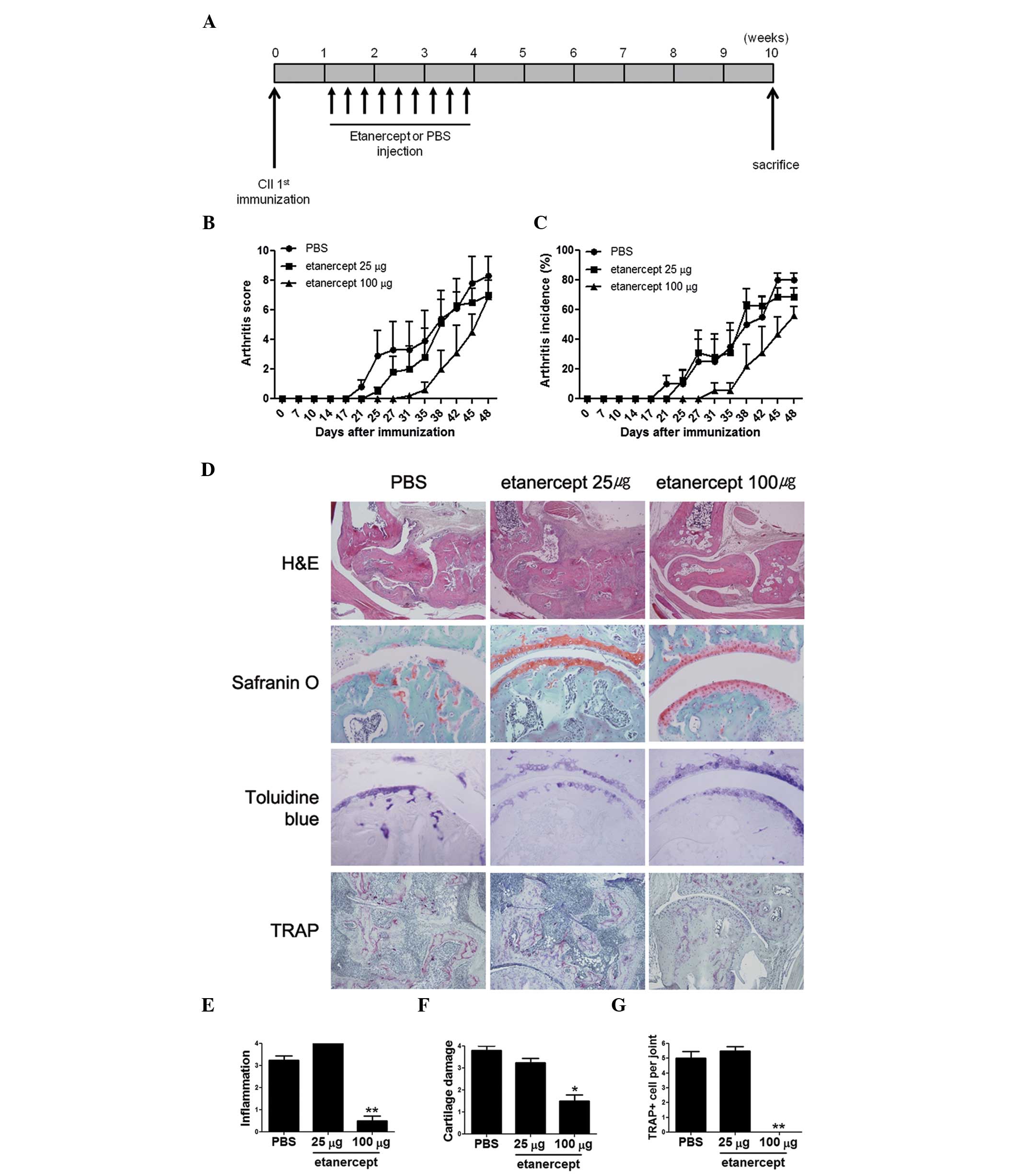
immunization (Fig. 1A)., although
the extent of inflammation commonly reaches a peak at week 8 in
this model of arthritis. Throughout this period, we monitored the
severity of arthritis in each group to examine whether etanercept
injection mitigates inflammation during the early stages of CIA,
and whether the degree of inflammation correlates with that of bone
loss. The results demonstrated that mice that were injected with
100 μg etanercept show a significant reduction in arthritic
symptoms compared to those injected with PBS (Fig. 1B and C), although the effects
appeared to last for only 18 days after the injections.
Demineralized bone was stained with H&E,
safranin O, and toluidine blue to determine whether
etanercept-mitigated inflammation resulted in a reduction of
cartilage erosion in the metacarpophalangeal joints of the animals
(Fig. 1D). The results showed that
mice treated with 100 μg etanercept display reduced infiltration
and cartilage erosion compared to control CIA mice, consistent with
the RA scores and incidence rates. Joint inflammation in the group
treated with 100 μg etanercept was ~1/7th of that observed in
control CIA mice, and cartilage damage was as low as 35% of that
observed in CIA mice (Fig. 1E and
F). Although the effects were not as significant as those seen
in mice treated with 100 μg etanercept, the group treated with 25
μg etanercept also showed a reduction in arthritis. In this case,
the degree of inflammation was slightly higher compared to control
CIA mice, but cartilage damage was reduced, indicating that 25 μg
of etanercept can prevent cartilage damage.
Since TNF-α is one of the key inducers of
osteoclastogenesis (17), we
assumed that etanercept treatment can reduce the number of
osteoclasts, thus reducing bone damage. The bones were stained with
TRAP to determine the number of multinucleated osteoclasts
(Fig. 1G). No osteoclast was
detected in the bones or joints of mice treated with 100 μg
etanercept, whereas a considerable number of osteoclasts were
detected in joints of control CIA mice, and of mice treated with 25
μg etanercept, consistent with the degree of cartilage damage in
these groups.
Anti-etanercept antibody production is
induced in mice challenged with high doses of etanercept
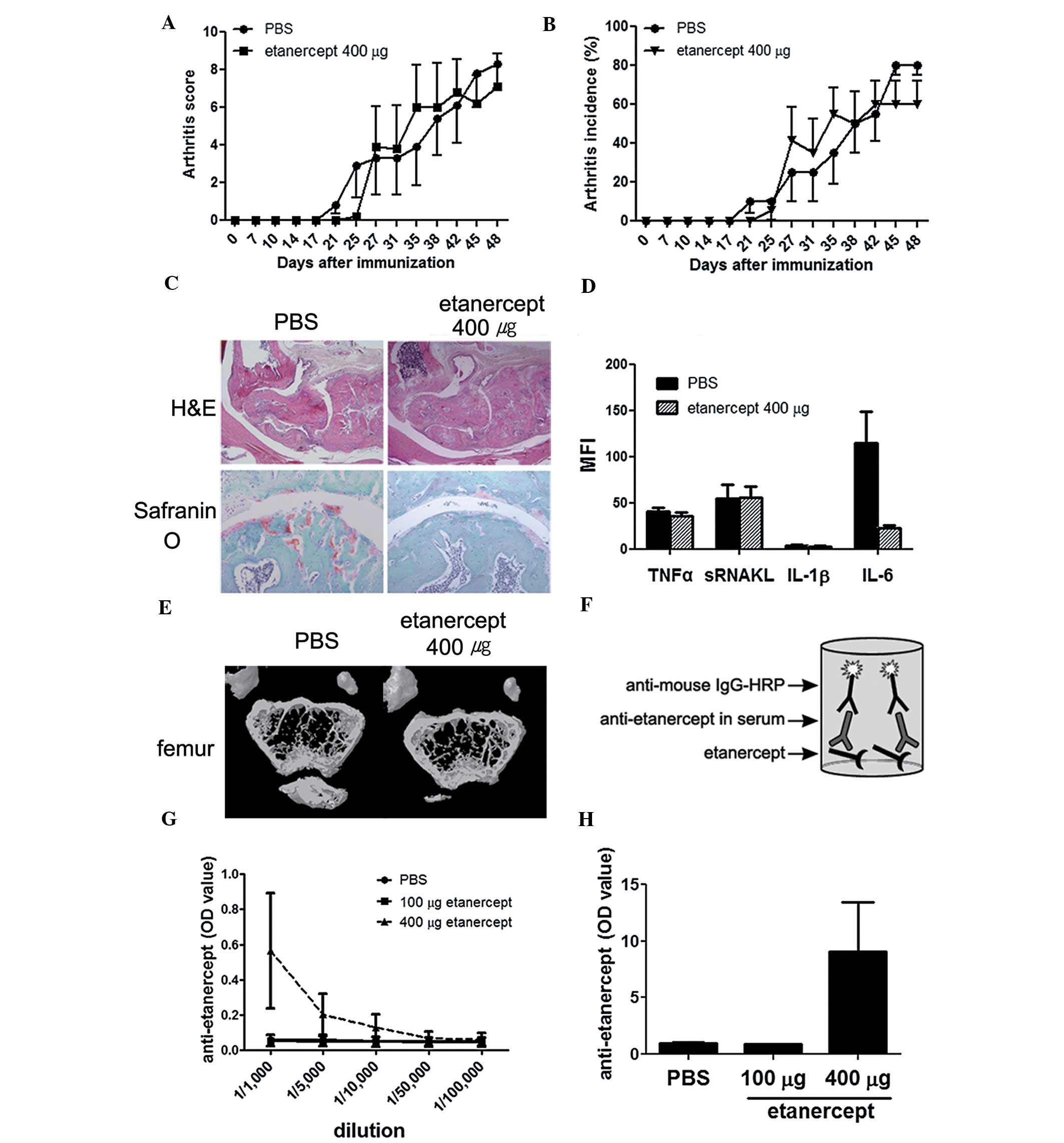
We performed the same experiments with a higher dose
of etanercept, 400 μg. The mice treated with this dose showed
little or no difference to PBS-treated mice (Fig. 2A–E). The levels of TNF-α and
soluble receptor activator of nuclear factor κ-B ligand (sRANKL) in
the serum of high-dose etanercept-treated mice were similar to
those measured in the serum of control CIA mice (Fig. 2D). Bone destruction and
demineralization were prominent in this group (Fig. 2E). We measured the concentration of
the anti-etanercept antibody at the time of sacrifice using ELISA
(Fig. 2F). Notably, the
anti-etanercept antibody was detected in mice treated with 400 μg
etanercept, even when serum samples were diluted by 1/1,000
(Fig. 2G and H).
Etanercept affects the level of
pro-inflammatory cytokines in the serum of CIA mice
Since TRAP staining data showed that etanercept
affects the number of osteoclasts in the ankle joints, we assumed
that there may be differences in the levels of cytokines related to
osteoclastogenesis in the sera of these mice. To examine whether
etanercept treatment up- or downregulates TNF-α, sRANKL,
interleukin (IL)-1β and IL-6, we collected sera from the orbital
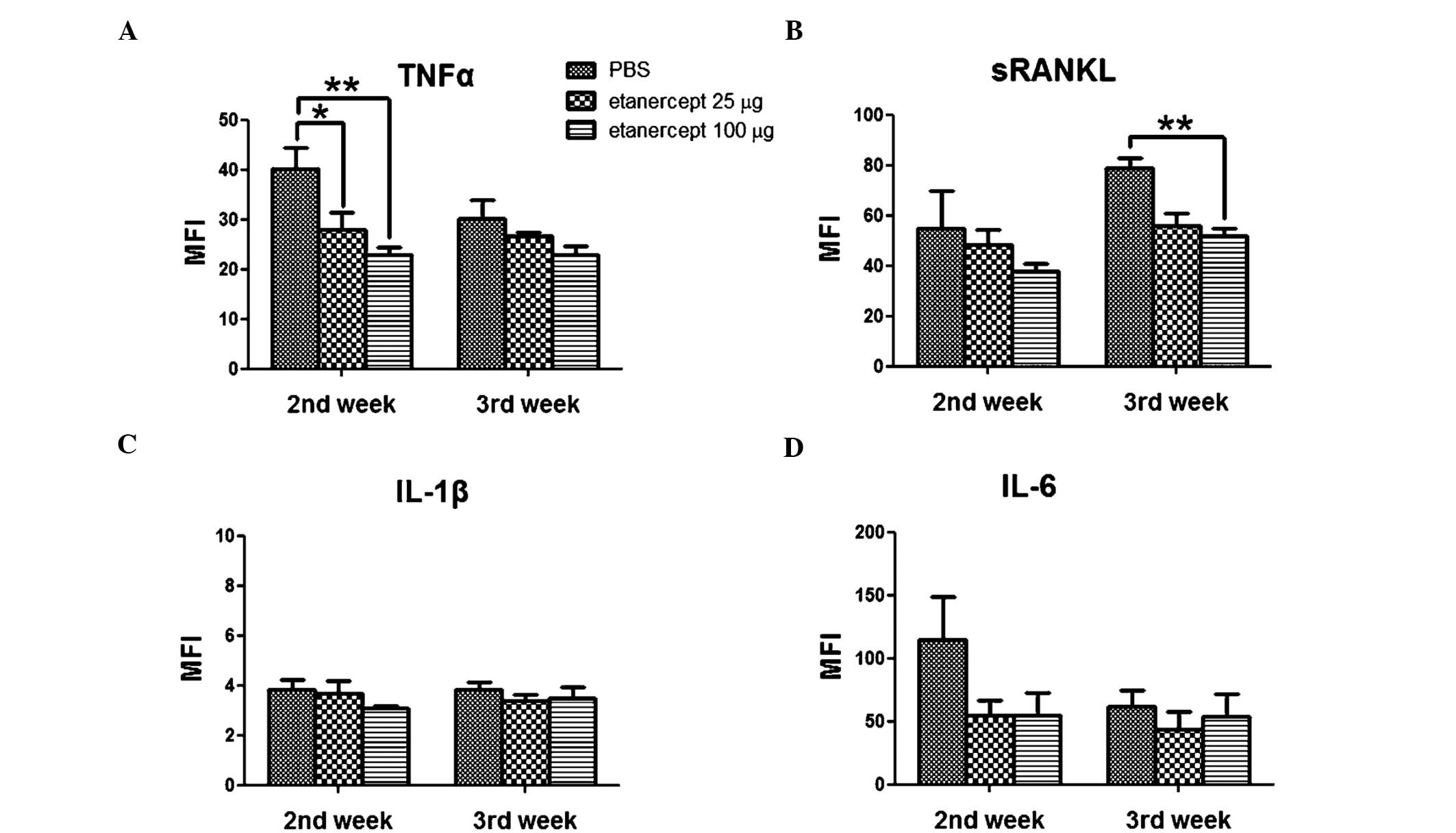
sinus of anesthetized animals at the second and third week after
the first collagen immunization. At 2 weeks after immunization, the
serum levels of TNF-α and sRANKL were consistent with the results
described in Fig. 1 (Fig. 3A and B). The lowest concentrations
of TNF-α and sRANKL were found in the sera of mice treated with 100
μg of etanercept. The dosages of 25, 100 and 400 μg were compared.
Among them 100 μg was more effective than 25 μg (Fig. 1), however, at 400 μg showed no
improvement (Fig. 2). TNF-α and
sRANKL levels in the serum of mice treated with 25 μg etanercept
were lower compared to those measured in the serum of control CIA
mice, but were higher compared to those measured in the serum of
mice that received the most effective dose (100 μg), indicating
dose dependency. The IL-6 levels showed time-dependent variation;
at the second week following the first collagen immunization, the
highest IL-6 levels were observed in the serum of control CIA mice,
but these levels were almost identical in all groups by the third
week. By contrast, the levels of IL-1β were relatively low
throughout the experiment and did not significantly differ between
groups (Fig. 3C and D). These data
showed that the TNF-α and sRANKL levels in the sera are largely
affected by etanercept treatment, and indicate that systemic bone
loss may be regulated by etanercept, through inhibition of
osteoclastogenesis.
Etanercept treatment at the early stage
of disease prevents systemic bone demineralization
As our data showed, etanercept reduced
osteoclastogenesis and affected the level of
osteoclastogenesis-associated cytokines, TNF-α and sRANKL. We next
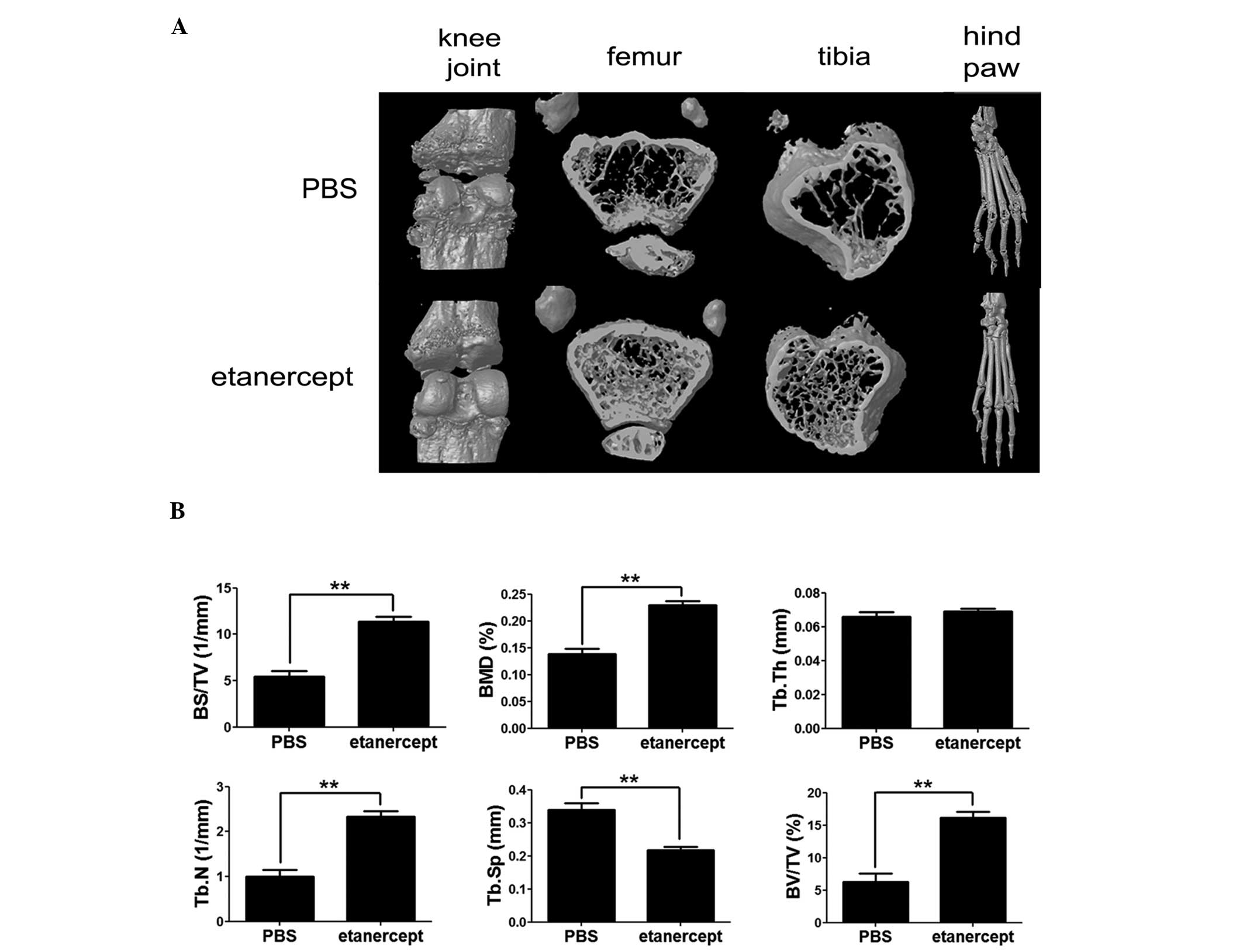
investigated, using micro-CT analysis, whether etanercept can
prevent systemic bone loss. The distal femoral and proximal tibial
metaphyses from each mouse were amputated and analyzed by micro-CT
to identify the effects of etanercept on bone demineralization,
induced by arthritis (Fig. 4A and
B). As expected, mice treated with a low dose of etanercept (25
μg) did not show any considerable improvement of disease symptoms,
and we therefore focused on mice treated with 100 μg etanercept.
Compared to the control CIA mice, the 100 μg etanercept-treated
group showed a 3-fold increase in the percentage of bone volume
(BV/TV), ~65% higher bone mineral density (BMD), nearly 2-fold
higher bone surface density (BS/TV), ~30% lower trabecular
separation (Tb.Sp), and a 2-fold higher trabecular number (Tb.N).
These results indicate that etanercept inhibits bone
demineralization in this group (Fig.
4A and B). The three-dimensional images of knee joints and hind
paws clearly illustrated this tendency (Fig. 4A). Trabecular thickness (Tb.Th) was
similar in all groups. These data indicate that etanercept may
attenuate systemic demineralization and focal erosion in CIA mice,
even when it is administered at the early stage of the disease.
Discussion
In this study, we showed that etanercept can
ameliorate both osteoporosis and inflammation in CIA mice. By
treating with etanercept only during the early stage of arthritis,
we measured how long the effect of etanercept can last and how
important anti-TNF-α treatment is, in an experimental RA model
(Fig. 1A). The anti-inflammatory
effect of 100 μg etanercept lasted ~18 days following cessation of
treatment (Fig. 1B and C). The
formation of pannus with bone erosion tended to correlate with the
severity of swelling, showing that 100 μg etanercept is effective
against both inflammation and focal bone erosion in RA (Fig. 1D–G).
We examined differences in the sera cytokine levels
of mice following etanercept treatment (Fig. 3). The results showed that sRANKL
and TNF-α decreased as the disease ameliorated, suggesting that the
effect of etanercept correlates with changes in these cytokines.
Both sRANKL and TNF-α are well-known mediators of
osteoclastogenesis; sRANKL combined with M-CSF is sufficient to
induce osteoclastogenesis and has been widely used in in
vitro osteoclast formation experiments (18–20).
Moreover, TNF-α stimulates osteoclast differentiation independently
of RANK-RANKL signaling (17).
Therefore, the reduction of sRANKL and TNF-α levels in mice treated
with 100 μg etanercept may inhibit osteoclastogenesis, preventing
the CIA-induced bone loss.
BMD was measured by micro-CT to investigate whether
the effect of etanercept is consistent with a systemic reduction in
bone erosion. Systemic bone destruction was reduced in the 100 μg
etanercept-treated mice compared to that in control CIA mice
(Fig. 4A and B), suggesting that
etanercept prevents not only destruction of focal bone adjacent to
the joints, but also systemic bone destruction. Furthermore, during
the early stage of the disease, these effects were observed only
after etanercept treatment, indicating that etanercept treatment
may be effective in preventing osteoporosis in patients with early
RA.
Notably, the highest dose of 400 μg etanercept did
not reduce the symptoms of arthritis. The incidence of arthritis
and the arthritis scores of mice treated with 400 μg etanercept
exceeded those of control CIA mice (Fig. 2A and B). Furthermore, pannus
formation and cartilage damage in the joints of mice treated with
400 μg etanercept were similar to those in CIA mice (Fig. 2C). The levels of sRANKL and TNF-α
in sera of mice treated with 400 μg etanercept did not decrease,
and no anti-osteoporotic effect was detected in these mice
(Fig. 2D and E).
Etanercept is considered effective for treating RA
and has been traditionally prescribed to many RA patients. Numerous
scientists and clinicians consider that an abnormal inhibition of
etanercept activity by several known factors can explain the fact
that some patients do not respond to this treatment. The
development of ADA against etanercept could be one of these
factors. However, etanercept was considered less likely to induce
development of ADA in comparison to other monoclonal antibody
drugs. Since the current literature does not unequivocally exclude
the possibility of induction of an anti-etanercept antibody, in the
present study we used an ELISA assay to detect this antibody
(Fig. 2F and H). We clearly
detected the anti-etanercept antibody only in the sera from the
etanercept-refractory group (400 μg). The recommended etanercept
dose for humans is 714 μg/kg; given that a mouse weighs 20 g, this
is equivalent to ~178.5 μg/mouse, considering the body surface area
(21). Thus, 400 μg of etanercept
was an overdose for the mice, which might have led to the synthesis
of the anti-etanercept antibody that may have caused the
ineffectiveness of the high-dose etanercept treatment. Although the
number of patients refractory to etanercept has continued to
increase (22), the reason for
this has not been elucidated. Several studies have suggested that
the production of anti-TNF-α inhibitor antibodies is the main
reason for treatment refractoriness (23–25).
However, the frequency of the anti-etanercept antibody was reported
at <5.6% (26–27), considerably lower than that of
other anti-TNF-α inhibitors (12–44% anti-infliximab and 6–87%
anti-adalimumab) (28), and no
correlation has been demonstrated between the presence of the
anti-etanercept antibody and poor clinical response. In this study,
we have shown that the production of anti-etanercept antibody by
mice exposed to an etanercept overdose is associated with the
inefficiency of the drug. We have shown that etanercept effectively
reduces inflammation and focal and systemic osteoporosis, and that
these effects partially result from the indirect inhibition of
osteoclastogenesis. These effects were considerable, even when
etanercept was only administered during the early stage of the
disease, suggesting that etanercept treatment during early stages
may be useful to ameliorate RA. We also demonstrated the presence
of anti-etanercept antibody in the serum of etanercept
treated-refractory mice, which provides a clue to the potential
mechanism of resistance in patients with RA who are refractory to
treatment.
Acknowledgements
We are very grateful to Karin for professional
proofreading. This study was supported by a grant of the Korea
Healthcare Technology R&D project, Ministry for Health, Welfare
& Family Affairs, Republic of Korea (A092258).
References
|
1
|
Aletaha D, Neogi T, Silman AJ, et al: 2010
rheumatoid arthritis classification criteria: an American College
of Rheumatology/European League Against Rheumatism collaborative
initiative. Ann Rheum Dis. 69:1580–1588. 2010. View Article : Google Scholar
|
|
2
|
Taylor PC: Pharmacology of TNF blockade in
rheumatoid arthritis and other chronic inflammatory diseases. Curr
Opin Pharmacol. 10:308–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Scott DL and Kingsley GH: Tumor necrosis
factor inhibitors for rheumatoid arthritis. N Engl J Med.
355:704–712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moreland LW, Schiff MH, Baumgartner SW, et
al: Etanercept therapy in rheumatoid arthritis. A randomized,
controlled trial Ann Intern Med. 130:478–486. 1999.PubMed/NCBI
|
|
5
|
Bathon JM, Martin RW, Fleischmann RM, et
al: A comparison of etanercept and methotrexate in patients with
early rheumatoid arthritis. N Engl J Med. 343:1586–1593. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moreland LW, Baumgartner SW, Schiff MH, et
al: Treatment of rheumatoid arthritis with a recombinant human
tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J
Med. 337:141–147. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Elliott MJ, Maini RN, Feldmann M, et al:
Randomised double-blind comparison of chimeric monoclonal antibody
to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid
arthritis. Lancet. 344:1105–1110. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van den Berg WB, Joosten LA and van de Loo
FA: TNF alpha and IL-1 beta are separate targets in chronic
arthritis. Clin Exp Rheumatol. 17:S105–S114. 1999.PubMed/NCBI
|
|
9
|
Kang KY, Lee KY, Kwok SK, et al: The
change of bone mineral density according to treatment agents in
patients with ankylosing spondylitis. Joint Bone Spine. 78:188–193.
2011. View Article : Google Scholar
|
|
10
|
Ju JH, Kang KY, Kim IJ, et al:
Visualization and localization of rheumatoid knee synovitis with
FDG-PET/CT images. Clin Rheumatol. 27(Suppl 2): S39–S41. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wolfe F and Michaud K: Towards an
epidemiology of rheumatoid arthritis outcome with respect to
treatment: randomized controlled trials overestimate treatment
response and effectiveness. Rheumatology (Oxford). 44 Suppl
4:iv18–iv22. 2005. View Article : Google Scholar
|
|
12
|
National Institute for Health and Clinical
Excellence. Adalimumab, etanercept and infliximab for the treatment
of rheumatoid arthritis. http://www.nice.org.uk/nicemedia/live/11867/37914/37914.pdf.
Accessed June 8, 2012
|
|
13
|
Genovese MC, Bathon JM, Martin RW, et al:
Etanercept versus methotrexate in patients with early rheumatoid
arthritis: two-year radiographic and clinical outcomes. Arthritis
Rheum. 46:1443–1450. 2002.PubMed/NCBI
|
|
14
|
O’Dell JR: Treating rheumatoid arthritis
early: a window of opportunity? Arthritis Rheum. 46:283–285.
2002.PubMed/NCBI
|
|
15
|
Ju JH, Cho ML, Jhun JY, et al: Oral
administration of type-II collagen suppresses IL-17-associated
RANKL expression of CD4+ T cells in collagen-induced
arthritis. Immunol Lett. 117:16–25. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yamaguchi N, Ohshima S, Umeshita-Sasai M,
et al: Synergistic effect on the attenuation of collagen induced
arthritis in tumor necrosis factor receptor I (TNFRI) and
interleukin 6 double knockout mice. J Rheumatol. 30:22–27.
2003.PubMed/NCBI
|
|
17
|
Kobayashi K, Takahashi N, Jimi E, et al:
Tumor necrosis factor alpha stimulates osteoclast differentiation
by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp
Med. 191:275–286. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hsu H, Lacey DL, Dunstan CR, et al: Tumor
necrosis factor receptor family member RANK mediates osteoclast
differentiation and activation induced by osteoprotegerin ligand.
Proc Natl Acad Sci USA. 96:3540–3545. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boyle WJ, Simonet WS and Lacey DL:
Osteoclast differentiation and activation. Nature. 423:337–342.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lacey DL, Timms E, Tan HL, et al:
Osteoprotegerin ligand is a cytokine that regulates osteoclast
differentiation and activation. Cell. 93:165–176. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reagan-Shaw S, Nihal M and Ahmad N: Dose
translation from animal to human studies revisited. FASEB J.
22:659–661. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Finckh A, Simard JF, Gabay C and Guerne
PA; SCQM physicians. Evidence for differential acquired drug
resistance to anti-tumour necrosis factor agents in rheumatoid
arthritis. Ann Rheum Dis. 65:746–752. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Haraoui B, Cameron L, Ouellet M and White
B: Anti-infliximab antibodies in patients with rheumatoid arthritis
who require higher doses of infliximab to achieve or maintain a
clinical response. J Rheumatol. 33:31–36. 2006.PubMed/NCBI
|
|
24
|
Elkayam O, Burke M, Vardinon N, et al:
Autoantibodies profile of rheumatoid arthritis patients during
treatment with infliximab. Autoimmunity. 38:155–160. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Atzeni F, Doria A, Ghirardello A, et al:
Organ-specific autoantibodies in patients with rheumatoid arthritis
treated with adalimumab: a prospective long-term follow-up.
Autoimmunity. 41:87–91. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dore RK, Mathews S, Schechtman J, et al:
The immunogenicity, safety, and efficacy of etanercept liquid
administered once weekly in patients with rheumatoid arthritis.
Clin Exp Rheumatol. 25:40–46. 2007.PubMed/NCBI
|
|
27
|
Keystone EC, Schiff MH, Kremer JM, et al:
Once-weekly administration of 50 mg etanercept in patients with
active rheumatoid arthritis: results of a multicenter, randomized,
double-blind, placebo-controlled trial. Arthritis Rheum.
50:353–363. 2004. View Article : Google Scholar
|
|
28
|
Emi Aikawa N, de Carvalho JF, Artur
Almeida Silva C and Bonfá E: Immunogenicity of anti-TNF-alpha
agents in autoimmune diseases. Clin Rev Allergy Immunol. 38:82–89.
2010.PubMed/NCBI
|