Introduction
Squamous cell carcinoma (SCC) is the most common
malignancy of the oral cavity accounting for at least 92.8% of all
oral malignancies (1).
Furthermore, it accounts for ~3% of all malignant tumors in humans.
Every year, >500,000 individuals are diagnosed with oral
squamous cell carcinoma (OSCC) worldwide (2). Over the last decade, despite improved
diagnostic and therapeutic strategies, the 5-year survival rate of
OSCC remains poor (3). Thus, the
development of effective diagnostic and therapeutic candidates is
urgently required.
Vasculogenesis and angiogenesis are important for
the formation of new blood vessels, which is crucial in the
development and progression of malignant tumors (4). This process is tightly regulated by
various factors, including epidermal growth factor-like domain 7
(EGFL7). EGFL7, also known as VE-statin, MEGF7 and Notch4-like
protein or Zneu1, was initially reported to inhibit the migration
of human aortic smooth muscle cells in vitro, indicating
that it may participate in vessel maturation (5). Further investigation revealed that
EGFL7 was an important tubulogenic factor in the process of
vasculogenesis (6). MicroRNA-126
(miR-126) is an endothelial-specific miRNA located within intron 7
of EGFL7 and has recently been found to be involved in the process
of blood vessel formation. MicroRNAs (miRNAs) represent a class of
~22 nucleotide long, non-coding RNAs, which have been identified as
important suppressive regulators of gene expression by the
inhibition of protein translation, and to a lesser extent by mRNA
degradation. Previously, accumulating evidence revealed the role of
miR-126 in multiple types of cancer. Generally, the expression
levels of miR-126 were downregulated in cancer tissues, while its
overexpression significantly inhibited tumor growth, migration and
invasiveness, partly through suppressing cancer cell proliferation.
However, the role of miR-126 as well as the exact regulatory
mechanism in OSCC remains to be fully elucidated.
In the present study, we examined the expression
level of miR-126 as well as EGFL7 in 20 OSCC tissues, their matched
adjacent tissues, as well as three OSCC cell lines, Tca8113,
OSCC-15 and CAL27. Furthermore, an miR-126 mimic and miR-126
inhibitor were used to examine the role of miR-126 in OSCC-15 cells
by examining cell proliferation, apoptosis, cell cycle
distribution, colony formation and invasion. Furthermore, we also
determined the effect of miR-126 overexpression and inhibition on
the secretion of vascular endothelial growth factor (VEGF) and
basic fibroblast growth factor (bFGF), respectively.
In conclusion, our data suggests that miR-126 acts
as a tumor suppressor in OSCC cells and thus may serve as a
promising diagnostic and therapeutic target for OSCC.
Materials and methods
Reagents and materials
Fetal bovine serum (FBS), TRIzol, TaqMan qRT-PCR
miRNA assay kit, SYBR-Green qPCR mix, Lipofectamine 2000, miR-126
mimics and an miR-126 inhibitor were purchased from Thermo Fisher
Scientific (Waltham, MA, USA). High glucose Dulbecco’s modified
Eagle’s medium (H-DMEM) was purchased from Gibco Laboratories
(Grand Island, NY, USA). Mouse anti-EGFL7 monoclonal antibody,
mouse anti-GAPDH monoclonal antibody and rabbit anti-mouse
secondary antibody were purchased from Abcam (Cambridge, UK). MTT
was purchased from Sigma (St. Louis, MO, USA). Propidium iodide
(PI) was purchased from Roche Molecular Biochemicals (Indianapolis,
IN, USA). The cell invasion assay kit was obtained from Corning
Inc. (Corning, NY, USA). The VEGF ELISA kit and bFGF ELISA kit were
purchased from R&D Systems (Minneapolis, MN, USA).
Tissue specimen collection
The present study was approved by the Ethics
Committee of Xiangya Medical College, Central South University
(Changsha, Hunan, China). Following informed consent being obtained
from each patient, 10 cases of fresh-frozen OSCC tissues and their
matched normal adjacent tissues, as well as 10 cases of normal
tissues were obtained from patients at the Department of
Stomatology, The Second Affiliated Xiangya Hospital of Central
South University (Changsha, Hunan, China) from January to June
2012. No patient had undergone radiotherapy or chemotherapy prior
to surgery. Following surgical removal, all samples were
immediately snap-frozen in liquid nitrogen and stored at −80°C
until use.
Cell culture
Three OSCC cell lines, Tca8113, OSCC-15 and CAL27,
were purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). All cells were cultured in DMEM supplemented
with 10% FBS, 100 IU/ml of penicillin and 100 μg/ml of streptomycin
sulfate at 37°C in a humidified incubator containing 5%
CO2. All experiments were performed at the third
passage.
RNA extraction and reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from tissues and cells using
TRIzol. RNA was synthesized to cDNA using the RT-PCR kit in
accordance with the manufacturer’s instructions. For the EGFL7
assay, SYBR-Green qPCR mix was used to perform qRT-PCR. The EGFL7
primer was as follows: forward 5′-TGAATGCAGTGCTAGGAGGG-3′ and
reverse 5′-GCACACAGAGTGTACCGTCT-3′. Glyceraldehyde phosphate
dehydrogenase (GAPDH) was used as an internal control with the
sense primer 5′-ACAACTTTGGTATCGTGGAAGG-3′ and antisense primer
5′-GCCATCACGCCACAGTTTC-3′. For the detection of miR-126 expression,
the TaqMan qRT-PCR miRNA assay kit was used to perform real-time
RT-PCR and analyzed with an ABI 7500 Sequence Detection System. U6
was used as an internal control. The relative expression levels of
genes were analyzed using the 2−ΔΔCt method. Independent
experiments were repeated three times.
Western blotting
Tissues or cells were solubilized in cold RIPA lysis
buffer. Protein (20 μg per lane) was separated with 12% SDS-PAGE.
Following that, the proteins were transferred onto nitrocellulose
membranes, which were then inhibited in 5% non-fat dried milk in
PBST for 3 h and then incubated overnight with mouse anti-EFGL7
monoclonal antibody (1:200) or mouse anti-GAPDH monoclonal antibody
(1:400). Following washing with PBS three times (each for 5 min),
the membranes were incubated with rabbit anti-mouse secondary
antibody (1:40,000) for 1 h at room temperature. Then, the ECL kit
(Huyu Co., Shanghai, China) was used to detect the immune
complexes. Following that, the membranes were scanned for the
relative value of protein expression in gray scale by Image-Pro
plus software 6.0. The relative expression level of protein is
presented as the density ratio versus GAPDH.
Transfection
Cells were cultured to ~70% confluence and then were
resuspended in serum-free H-DMEM at a concentration of 100,000
cells/ml. Six-well plates were used to inoculate with 2 ml
suspension for each well. According to the manufacturer’s
instructions, mir-126 mimics, NC mimics or an miR-126 inhibitor was
diluted with 250 μl of serum-free H-DMEM. Lipofectamine 2000
transfection reagent (50 μl) was diluted with 2.5 ml of serum-free
H-DMEM. Then, the diluted Lipofectamine 2000 transfection reagent
was added into the mimics or inhibitor dilution, mixed gently and
incubated for 20 min at room temperature. The cell suspension was
washed with serum-free H-DMEM two times, added with the mixture and
then incubated at 37°C and 5% CO2 for 6 h. Following
that, the medium in each well was replaced by the normal
serum-containing medium and cultured for 24 h prior to the
following experiments.
Cell proliferation assay
For each group, 10,000 cells per well were plated in
a 96-well plate. Then, the plates were incubated for 12, 24, 48 or
72 h at 37°C and 5% CO2. The MTT assay was performed to
determine the cell proliferation. Each well was added with 25 μl of
MTT (10 mg/ml) and then incubated for 4 h at 37°C and 5%
CO2. Then, the supernatant was removed and each well was
added with 150 μl of DMSO. The absorbance was detected at 570 nm
with a Microplate Reader (Bio-Rad, Hercules, CA, USA). Each assay
was performed in triplicate wells.
Cell cycle distribution assay
At 48 h after transfection, cells were fixed with
70% ethanol. Then, the cells were stained with 25 lg/ml of PI in
PBS containing 0.1% BSA, 0.05% Triton X-100 and 50 lg/ml of RNaseA
for 30 min at room temperature. Following that, the cells were
analyzed by flow cytometry.
Colony formation assay
For each group, each well of a 6-well plate was
added with 3 ml of complete medium containing 150 cells and then
incubated at 37°C and 5% CO2 for 14 days. Then, cells
were gently washed and stained with Giemsa. Colonies containing at
least 50 cells were counted and images were captured.
Cell invasion assay
The cell invasion assays were performed in a 24-well
transwell chamber pre-coated with Matrigel. Cells were collected
and resuspended in serum-free H-DMEM at a concentration of 10,000
cells/ml, respectively. The upper chamber was added with 0.2 ml
cell suspensions. The bottom chamber was filled with 0.5 ml of
H-DMEM containing 10% FBS. Following incubation for 24 h at 37°C
and 5% CO2, a cotton bud was used to remove the cells
which were not through the polycarbonate membrane. Then, the cells
which moved through the polycarbonate membrane and adhered to the
bottom of it, were stained with Trypan blue for 15 min, and then
images were captured and the cells were counted.
Enzyme-linked immunosorbent assay
(ELISA)
Cell supernatants in each group were used to
determine the secretion of VEGF and bFGF using ELISA. The VEGF and
bFGF ELISA kits were used, according to the manufacturer’s
instructions, and the concentrations of VEGF and bFGF were
calculated. Optical density (OD) values were determined by a
microplate reader (Bio-Rad).
Statistical analysis
All data are expressed as the mean ± SD of three
independent experiments. Statistical analysis was performed using
SPSS.17 software. Statistical analysis of differences was performed
by one-way analysis of variance (ANOVA). P<0.05 was considered
to indicate a statistically significant difference
(**P<0.05, *P<0.01).
Results
Downregulation of miR-126 expression in
OSCC tissues and cell lines
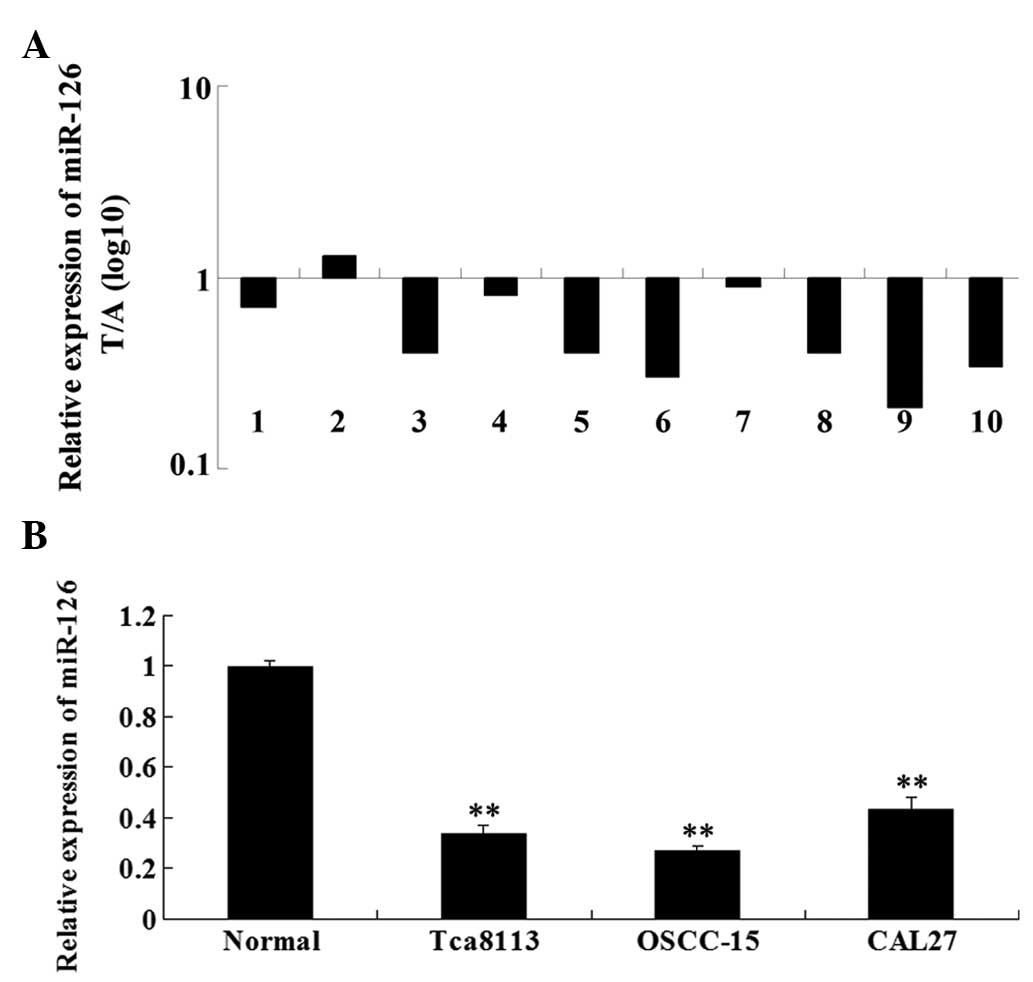
Firstly, we examined the expression level of miR-126
in 10 cases of OSCC samples and their matched adjacent tissues by
performing real-time RT-PCR. As shown in Fig. 1A, the expression of miR-126 was
significantly downregulated in OSCC tissues when compared with that
in the matched normal tissues. We further determined the miR-126
expression level in three OSCC cell lines, Tca8113, OSCC-15 and
CAL27. Our data demonstrated that the expression of miR-126 was
also downregulated in three OSCC cell lines compared with the
normal adjacent tissues (Fig. 1B).
These findings indicate that miR-126 downregulation may be involved
in the development of OSCC.
Upregulation of EGFL7 expression in OSCC
tissues and cell lines
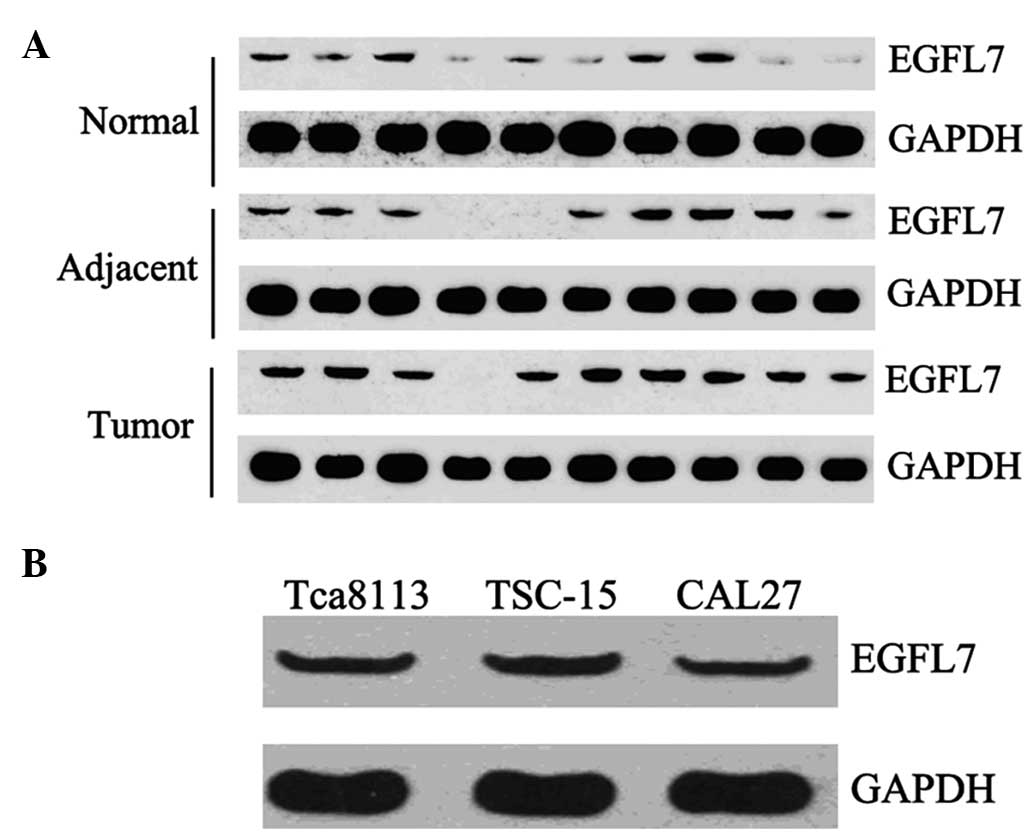
We further examined the protein level of EGFL7 in 10
cases of OSCC tissues and their matched adjacent tissues, as well
as 10 cases of normal tissues. Western blot analysis data
demonstrated that the expression of EGFL7 was notably increased in
OSCC tissues compared with their matched adjacent and normal
tissues (Fig. 2A). We also
determined the EGFL7 protein level in Tca8113, OSCC-15 and CAL27
cells. As shown in Fig. 2B, all
three cell lines demonstrated a positive protein expression of
EGFL7.
miR-126 negatively regulates the protein
expression of EGFL7 in OSCC-15 cells
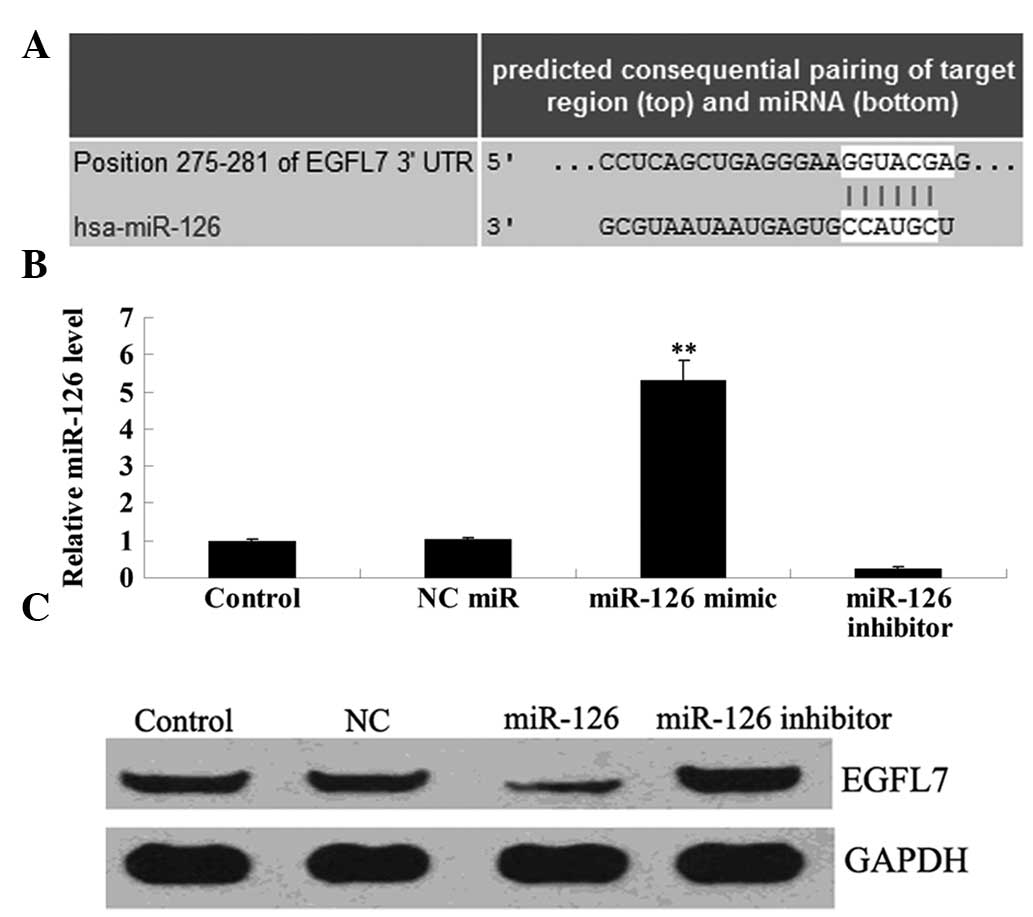
Since bioinformatical analysis data demonstrated
that EGFL7 was a putative target of miR-126, we preliminarily
investigated their relationship in OSCC-15 cells. Following
transfection with the miR-126 mimic and miR-126 inhibitor into
OSCC-15 cells, respectively, real-time PCR was performed to
determine the transfection efficiency. As shown in Fig. 3A, the expression level of miR-126
was markedly upregulated following transfection with the miR-126
mimic, and notably downregulated following transfection with the
miR-126 inhibitor, when compared with that in the control group,
respectively. Following confirmation of the transfection
efficiency, we applied western blotting to examine the protein
expression of EGFL7 in each group. As demonstrated in Fig. 3B, the expression level of EGFL7 was
reduced following transfection with the miR-126 mimic in OSCC-15
cells. By contrast, the EGFL7 expression level was upregulated in
OSCC-15 cells transfected with the miR-126 inhibitor. These
findings suggest that miR-126 negatively regulates the protein
expression of EGFL7 in OSCC-15 cells.
Effects of miR-126 upregulation and
downregulation on OSCC-15 cell proliferation
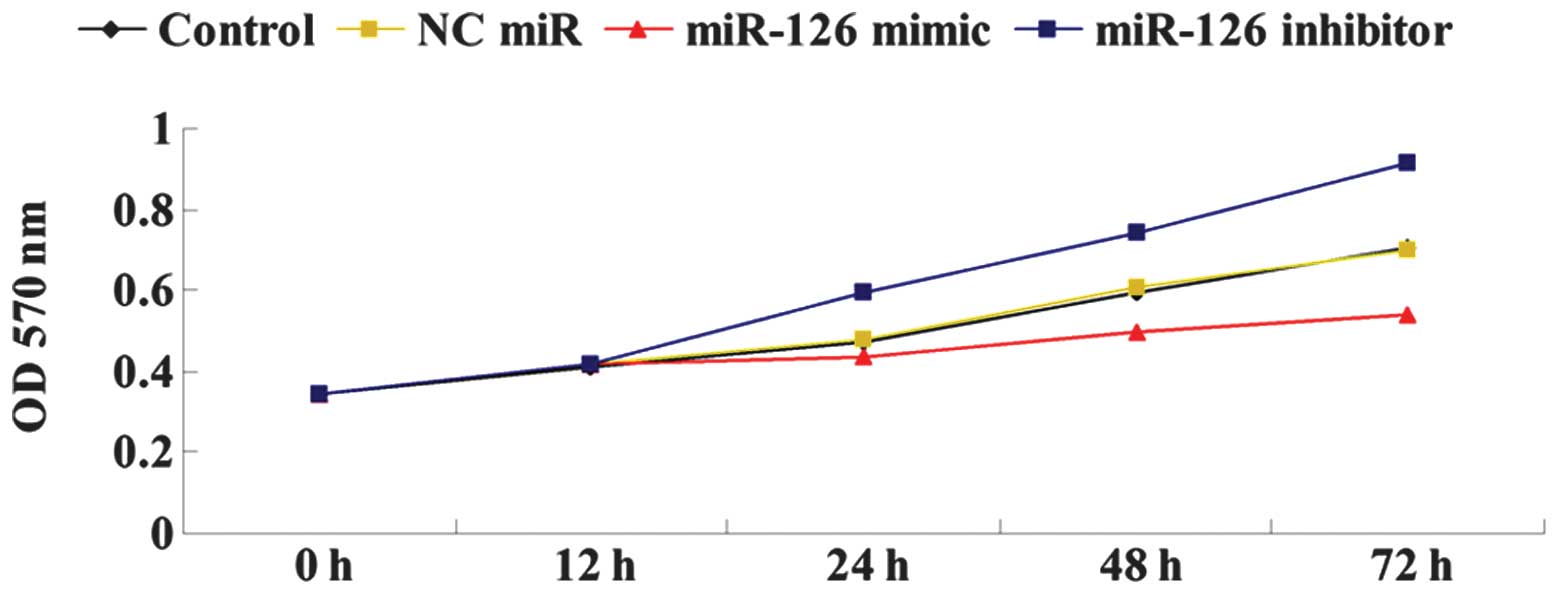
We further determined the effects of miR-126
overexpression or inhibition on OSCC-15 cell proliferation. As
shown in Fig. 4, the
overexpression of miR-126 significantly downregulated the cellular
proliferation of OSCC-15 cells, while the downregulation of miR-126
by its inhibitor markedly upregulated OSCC-15 cell proliferation.
Accordingly, we suggested that miR-126 has an inhibitory effect on
OSCC-15 cell proliferation.
Role of miR-126 in OSCC-15 cell
apoptosis
We investigated the role of miR-126 in OSCC-15 cell
apoptosis. As demonstrated in Fig.
5, the transfection of OSCC-15 cells with the miR-126 mimic
significantly upregulated their apoptosis, which could be reversed
by transfection with the miR-126 inhibitor. Thus, we suggested that
miR-126 can induce cell apoptosis in OSCC-15 cells.
Effect of miR-126 on the cell cycle
progression of OSCC-15 cells
To further investigate the effect of miR-126 on the
cell cycle progression of OSCC-15 cells, a cell cycle assay was
performed. Our findings demonstrated that the miR-126 overexpressed
OCSS-15 cells demonstrated the highest percentage in the G1 stage,
however, the lowest percentage in the S stage, when compared with
that in other groups, indicating that mitosis was inhibited in the
G1 stage. By contrast, OCSS-15 cells transfected with the miR-126
inhibitor demonstrated the highest percentage in the G2/M stage,
indicating that the inhibition of miR-126 promoted mitosis
(Fig. 6). These findings suggest
that miR-126 has a negative effect on cell cycle progression in
OCSS-15 cells.
Role of miR-126 in the regulation of cell
invasion in OSCC-15 cells
The cell invasion ability of OSCC-15 cells was
examined following transfection with the miR-126 mimic and
inhibitor, respectively. As shown in Fig. 7, miR-126 overexpression markedly
inhibited OSCC-15 cell invasion, while miR-126 inhibition
significantly promoted the invasion of OSCC-15 cells, when compared
with that in the control groups.
Effects of miR-126 upregulation and
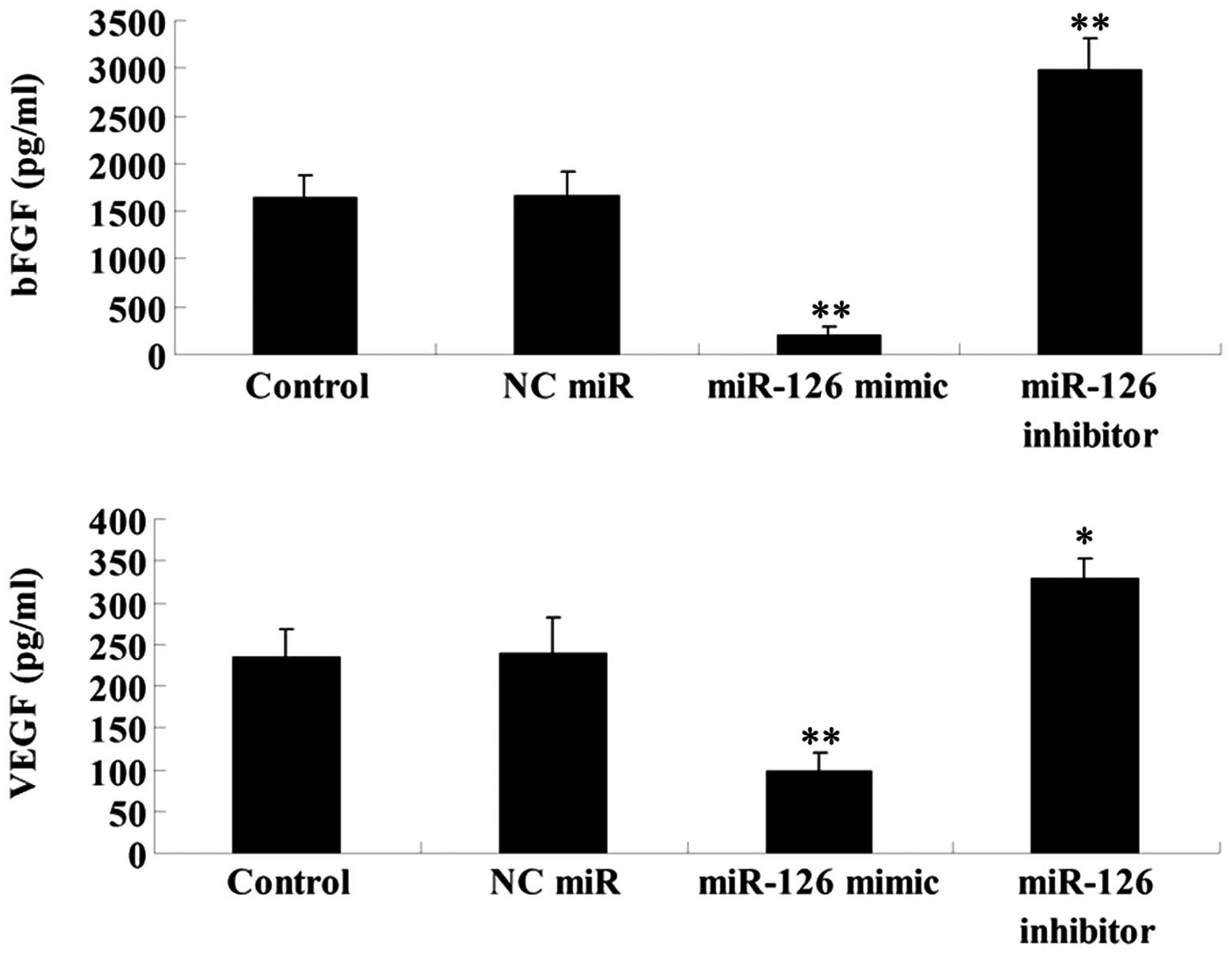
inhibition on the VEGF and bFGF secretion in OSCC-15 cells
As EGFL7 has been demonstrated to be implicated in
the regulation of angiogenesis, we performed ELISA to determine the
secretion levels of two vascular markers VEGF and bFGF in OSCC-15
cells transfected with the miR-126 mimic and inhibitor,
respectively. Our findings demonstrated that miR-126 overexpression
significantly downregulated the secretion of VEGF and bFGF in
OSCC-15 cells, while miR-126 inhibition markedly promoted their
secretion (Fig. 8). These findings
suggest that miR-126 may possess an inhibitory function in
angiogenesis, partially at least via suppressing EGFL7
expression.
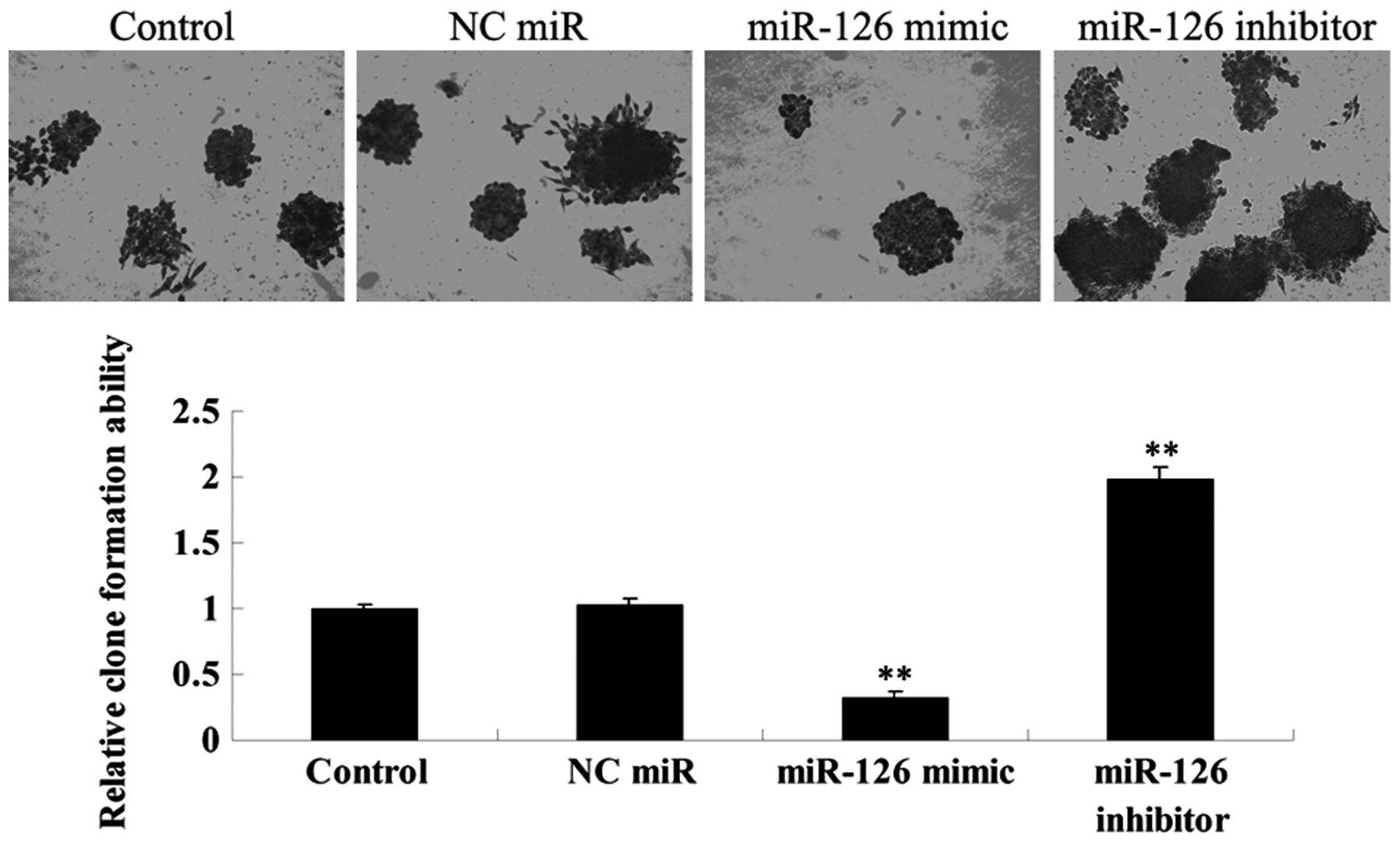
Effects of miR-126 upregulation and
downregulation on the colony-formation efficiency of OSCC-15
cells
The effects of miR-126 overexpression and inhibition
on colony-formation efficiency in OSCC-15 cells were investigated.
As demonstrated in Fig. 9, the
miR-126 overexpressed OSCC-15 cells demonstrated the lowest
colony-formation efficiency, while the miR-126 downregulated
OSCC-15 cells demonstrated the highest colony-formation efficiency,
when compared with that in the controls.
Discussion
miR-126 has been revealed to be important in
different physiological and pathological processes, including
inflammation (7), blood vessel
formation (8) as well as the
development and progression of malignant tumors (9). Altered expression of miR-126 has been
reported in various cancer cells, including breast cancer, colon
cancer, prostate cancer, lung cancer, cervical cancer, bladder
cancer, prostate cancer, gastric cancer and leukemia (9–16).
However, the function of miR-126 in OSCC cells remains largely
unknown. The present study suggests that miR-126 has a suppressive
effect on OSCC cells, mainly through inhibiting cell proliferation,
cell cycle progression, cell invasion and colony formation, as well
as inducing cell apoptosis. Furthermore, miR-126 upregulation
reduced VEGF and bFGF secretion, which are critical drivers for
angiogenesis and subsequent tumor growth (17). These findings indicate that miR-126
acts as a tumor suppressor in OSCC cells.
Recently, an increasing body of evidence has focused
on the role of miR-126 in various types of cancer. Guo et al
demonstrated that miR-126 inhibited colon cancer possibly via the
phosphatidylinositol 3-kinase signaling pathway (18). Liu et al reported that
miR-126 overexpression induced cell cycle arrest in vitro
and inhibited tumor cell growth in vivo, partially through
negatively targeting VEGF-A in lung carcinoma cells (19). In fact, the expression level of
miR-126 was also regulated by oncogenes. For instance, Li et
al reported that overexpression of the oncogene Src caused a
decrease in miR-126 expression in mouse embryonic Cx43KO brain
cells and the Src kinase inhibitor PP2 led to an increase in
miR-126 expression, which further decreased cell migration in
breast cancer cells (20).
Furthermore, miR-126 was found to directly target the 3′-UTR of
Crk, the downregulation of which leads to the inhibition of tumor
cell proliferation and invasion, through affecting the focal
adhesion network involved in integrin signaling (16,21).
Accordingly, miR-126 has been demonstrated to be involved in
multiple molecular signaling cascades in various cancer cells.
Furthermore, miR-126 is an intron-located miRNA and
its host gene is EGFL7. Generally, the expression of the majority
of the intron-located miRNA is paralleled by the transcription of
mRNAs encoded by the host genes. Recently, however, certain
regulatory elements upstream of the host genes were identified to
promote intron-located miRNA transcription independently. Thus, an
independent expression of miRNAs and mRNAs may also exist. Indeed,
one study previously reported that the expression of miR-126 was
regulated independently of EGFL7 (20), indicating that a separate promoter
may exist, driving miR-126 transcription. However, whether or not
this promoter element exists remains to be elucidated by further
investigations. In the present study, we demonstrated that the
expression of miR-126 negatively regulated the protein level of
EGFL7 and bioinformatical analysis data also demonstrated that
EGFL7 was a direct target of miR-126. These findings suggest that
although the expression pattern of miR-126 and EGFL7 remains
controversial, miR-126 is likely to inhibit EGFL7 expression at a
post-transcriptional level.
Furthermore, several studies have indicated that
miR-126 can promote endothelial cell proliferation and migration
(8,22) and thus may be essential for
angiogenesis. In theory, this putative function is counterintuitive
for its suppressive role in various types of cancer. However, the
exact role of miR-126 in tumor angiogenesis remains largely
uncovered. To connect the role of miR-126 in cancer cells to its
physiological function in the vasculature, we examined the effect
of miR-126 upregulation and downregulation on the secretion of VEGF
and bFGF, two key regulators for angiogenesis as well as cancer
development, and found that miR-126 had an inhibitory effect on
their production. Since blood vessel growth represents a key
feature in the pathogenesis of cancer, further investigation
regarding the effect of miR-126 on the tumor vasculature and the
surrounding vessels can help reveal its exact role in cancer
progression.
In summary, the present study suggests that miR-126
has a suppressive effect in OSCC cells and thus may become a
promising candidate for the development of a therapeutic strategy
for OSCC treatment.
References
|
1
|
Lo Muzio L, Santarelli A, Panzarella V, et
al: Oral squamous cell carcinoma and biological markers: an update
on the molecules mainly involved in oral carcinogenesis. Minerva
Stomatol. 56:341–347. 2007.PubMed/NCBI
|
|
2
|
Fan S, Tang QL, Lin YJ, et al: A review of
clinical and histological parameters associated with contralateral
neck metastases in oral squamous cell carcinoma. Int J Oral Sci.
3:180–191. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shi Z and Stack MS: Molecules of cell
adhesion and extracellular matrix proteolysis in oral squamous cell
carcinoma. Histol Histopathol. 25:917–932. 2010.PubMed/NCBI
|
|
4
|
Albini A, Tosetti F, Li VW, Noonan DM and
Li WW: Cancer prevention by targeting angiogenesis. Nat Rev Clin
Oncol. 9:498–509. 2012. View Article : Google Scholar
|
|
5
|
Soncin F, Mattot V, Lionneton F, et al:
VE-statin, an endothelial repressor of smooth muscle cell
migration. EMBO J. 22:5700–5711. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Parker LH, Schmidt M, Jin SW, et al: The
endothelial-cell-derived secreted factor Egfl7 regulates vascular
tube formation. Nature. 428:754–758. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harris TA, Yamakuchi M, Ferlito M, Mendell
JT and Lowenstein CJ: MicroRNA-126 regulates endothelial expression
of vascular cell adhesion molecule 1. Proc Natl Acad Sci USA.
105:1516–1521. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang S, Aurora AB, Johnson BA, et al: The
endothelial-specific microRNA miR-126 governs vascular integrity
and angiogenesis. Dev Cell. 15:261–271. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meister J and Schmidt MH: miR-126 and
miR-126*: new players in cancer. ScientificWorldJournal.
10:2090–2100. 2010.PubMed/NCBI
|
|
10
|
Wang X, Tang S, Le SY, et al: Aberrant
expression of oncogenic and tumor-suppressive microRNAs in cervical
cancer is required for cancer cell growth. PLoS One. 3:e25572008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crawford M, Brawner E, Batte K, et al:
MicroRNA-126 inhibits invasion in non-small cell lung carcinoma
cell lines. Biochem Biophys Res Commun. 373:607–612. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ladeiro Y, Couchy G, Balabaud C, et al:
MicroRNA profiling in hepatocellular tumors is associated with
clinical features and oncogene/tumor suppressor gene mutations.
Hepatology. 47:1955–1963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Z, Lu J, Sun M, et al: Distinct
microRNA expression profiles in acute myeloid leukemia with common
translocations. Proc Natl Acad Sci USA. 105:15535–15540. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saito Y, Friedman JM, Chihara Y, Egger G,
Chuang JC and Liang G: Epigenetic therapy upregulates the tumor
suppressor microRNA-126 and its host gene EGFL7 in human cancer
cells. Biochem Biophys Res Commun. 379:726–731. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Musiyenko A, Bitko V and Barik S: Ectopic
expression of miR-126*, an intronic product of the
vascular endothelial EGF-like 7 gene, regulates prostein
translation and invasiveness of prostate cancer LNCaP cells. J Mol
Med (Berl). 86:313–322. 2008.
|
|
16
|
Feng R, Chen X, Yu Y, et al: miR-126
functions as a tumour suppressor in human gastric cancer. Cancer
Lett. 298:50–63. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bremnes RM, Camps C and Sirera R:
Angiogenesis in non-small cell lung cancer: the prognostic impact
of neoangiogenesis and the cytokines VEGF and bFGF in tumours and
blood. Lung Cancer. 51:143–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo C, Sah JF, Beard L, Willson JK,
Markowitz SD and Guda K: The noncoding RNA, miR-126, suppresses the
growth of neoplastic cells by targeting phosphatidylinositol
3-kinase signaling and is frequently lost in colon cancers. Genes
Chromosomes Cancer. 47:939–946. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu B, Peng XC, Zheng XL, Wang J and Qin
YW: MiR-126 restoration down-regulate VEGF and inhibit the growth
of lung cancer cell lines in vitro and in vivo. Lung Cancer.
66:169–175. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li X, Shen Y, Ichikawa H, Antes T and
Goldberg GS: Regulation of miRNA expression by Src and contact
normalization: effects on nonanchored cell growth and migration.
Oncogene. 28:4272–4283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Birge RB, Kalodimos C, Inagaki F and
Tanaka S: Crk and CrkL adaptor proteins: networks for physiological
and pathological signaling. Cell Commun Signal. 7:132009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fish JE, Santoro MM, Morton SU, et al:
miR-126 regulates angiogenic signaling and vascular integrity. Dev
Cell. 15:272–284. 2008. View Article : Google Scholar : PubMed/NCBI
|