Introduction
The development and growth of a tumor requires a
sufficient blood supply. It is a widely accepted paradigm that
tumor vasculature is mostly generated through angiogenesis, the
process of new vessel growth from pre-existing vessels (1). However, tumors do not only rely on
host blood vessels for nourishment; they are also able to form
their own vasculature (2). In
1999, Maniotis et al (3)
reported that highly aggressive uveal melanomas are able to form
vessels using tumor cells instead of endothelial cells and this
phenomenon of tumor vascularization has been named vasculogenic
mimicry (VM). VM has been described in prostate cancer and certain
other types of highly aggressive tumors, and is associated with
tumor cell migration and invasion (4–7).
Several key molecules, including vascular endothelial
(VE)-cadherin, matrix metalloproteinases (MMPs), laminin-5γ2 chains
and CD147 have been implicated in VM (8,9).
Several studies have demonstrated that vascular endothelial growth
factor (VEGF) is important in the formation of VM and suggested
that VEGF→ephrin type-A receptor (EphA2)→MMPs→VM is the main
pathway for VM formation (10).
Although VM has been demonstrated to occur in prostate cancer, a
degree of which correlates with a poor clinical outcome (11), the precise molecular events
underlying the process of VM in prostate cancer have remained to be
fully elucidated.
Peroxisome proliferator-activated receptor (PPAR) is
a member of the family of nuclear receptor transcription factors.
PPARγ is expressed in numerous different tissues and regulates
lipid metabolism, glucose homoeostasis and inflammation (12). PPARγ is the molecular target of the
thiazolidinedione (TZD) class of antidiabetic drugs. The activation
of PPARγ by TZDs and other ligands have been demonstrated to
inhibit proliferation and invasion, and to induce apoptosis and
cell cycle arrest in prostate and other cancer cells through
PPARγ-dependent and -independent pathways (13–15).
Certain studies demonstrated that TZDs are able to induce the
inhibition of angiogenesis in cancer and human umbilical vein
endothelial cells (16,17). By contrast, several studies
suggested that PPARγ activation may enhance tumor angiogenesis
(18). In summary, the molecular
mechanisms underlying the anticancer effects of TZD remain
elusive.
Previous studies have demonstrated that the
activation of PPARγ affects angiogenesis and VEGF production by
tumor cells. In addition, VEGF is crucial in tumor angiogenesis and
VM formation (10). Thus, the
present study hypothesized that VM formation may be a potential
target of PPARγ ligands. The present study investigated the effect
of rosiglitazone (RSG), a ligand of PPARγ, on the formation of VM
in PC-3 cells and the underlying mechanisms in vitro. To the
best of our knowledge, the present study was the first to provide
evidence that exposure of PC-3 cells to RSG induced decreased
migration, invasion and formation of VM. The inhibitory effect of
RSG on VM formation may at least partially be explained by an
RSG-driven downregulation of VEGF and the phosphorylation of
AKT.
Materials and methods
Cell culture
The human prostate cancer cell line PC-3 was
maintained in RPMI-1640 medium (HyClone, Logan, UT, USA)
supplemented with 10% fetal bovine serum (FBS) (Gibco-BRL, Grand
Island, NY, USA), 1×105 U/l of penicillin and
streptomycin (HyClone, Logan, UT, USA) at 37°C in an incubator with
5% CO2.
Three-dimensional cultures
Matrigel® (BD Biosciences, San Jose, CA,
USA) was dropped onto glass coverslips and allowed to polymerize
for 1 h at 37°C. The cells were then seeded on top of the gels at a
high density and allowed to incubate. The addition of conditioned
media containing 10% FBS was performed by the pretreatment and the
continuous treatment regimes during the 48 h incubation period in
three-dimensional cultures (19).
Fresh media containing drugs was added every 24 h. For quantization
of VM networks, the number of tube connections per field
(magnification, ×40) was counted. The Student’s t-test was
performed on at least five fields for each variable collected from
three different experiments.
Cell migration assay
Cell migration was evaluated using an in
vitro wound-healing assay. PC-3 cells were plated in a six-well
plate and allowed to grow to confluence. The monolayer culture was
then scrape-wounded with a sterile micropipette tip to create a
denuded zone (gap) of constant width. The cells were exposed to
various concentrations of RSG, 24 h after removing the cellular
debris with phosphate-buffered saline (PBS). The speed of wound
closure was monitored by measuring the ratio of the distance to
that at 0 h. Each experiment was performed in triplicate and the
results are presented as the mean ± standard error.
Cell invasion assay
The invasive potential was quantified using a
Matrigel-coated transwell system, as previously described (20). The chamber (Corning Inc., Corning,
NY, USA), which contained an 8-μm pore size polycarbonate membrane
filter, was coated with Matrigel® and inserted into a 24
well culture plate. RSG was pre-incubated with PC-3 cells at
various concentrations, respectively, for 24 h prior to seeding.
The cell suspension (200 μl) was added into the upper chamber in
serum-free medium and 500 μl of medium, which was supplemented with
20% FBS, was added into the lower chamber. Following 48 h
incubation, the cells on the upper surface of the filters were
completely removed by wiping with cotton swabs. Then cells that
invaded the lower surface of the membrane were fixed with methanol
and stained with crystal violet. Each experiment was conducted in
triplicate. The cells were counted in five fields in each well by
light microscopy. The cells were assessed for their relative
invasion ability as percentages of the untreated controls.
Western blot analysis
Western blot analysis was performed to assess the
protein levels of VE-cadherin and the expression and
phosphorylation of signal transduction molecules, including AKT.
Following being treated with RSG (Sigma-Aldrich, St. Louis, MO,
USA), recombinant human (rh) VEGF165 (R&D Systems, Minneapolis,
MN, USA), LY294002 (Cell Signaling Technology, Danvers, MA, USA) or
GW9662 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), PC-3
cells were washed twice with PBS and treated with extraction buffer
(50 mM of Tris-Cl; pH 7.5; 150 mM of NaCl, 0.1% SDS, 1% NP-40 and
0.5% deoxycholic acid). The cell extractions were collected and
centrifuged at 10,000 × g for 10 min at 4°C, and the supernatants
were collected as cell lysates. The cell lysates were subjected to
SDS-PAGE and transferred onto polyvinylidene fluoride membranes
(Millipore, Bedford, MA, USA). The membranes were inhibited with 5%
(w/v) bovine serum albumin in Tris-buffered saline containing 0.1%
Tween-20 and then blotted with primary antibody. Subsequently, the
membranes were incubated with an appropriate secondary antibody
(horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit
immunoglobulin G; Wuhan Boster Biological Technology, Ltd, Wuhan,
Hubei, China). The immune-detected proteins were then visualized by
enhanced chemiluminescence. The untreated cells served as a
negative controls and β-actin was used as a control for equal
loading. The antibodies against VE-cadherin, AKT, p-AKT (Ser473)
and β-actin were purchased from Cell Signaling Technology (Beverly,
MA, USA).
ELISA assay
Conditioned medium was prepared as mentioned above
and VEGF concentrations in the medium were determined using a
commercial Human VEGF Quantikine ELISA kit (R&D Systems)
according to the manufacturer’s instructions. The absorbance at 450
nm was measured and corrected using the 540 nm reading on an
ELX-800 uv microplate reader (Bio-Tek Instruments, Inc, Winooski,
VT, USA).
quantitative polymerase chain reaction
(qPCR) assay
Total RNA was extracted using TRIzol reagent
according to the manufacturer’s instructions (Invitrogen Life
Technologies, Carlsbad, CA, USA). RNA (1 μg) was
reverse-transcribed. qPCR was accomplished with TransStart Green
qPCR SuperMix (Beijing TransGen Biotech Co., Ltd., Beijing, China)
using a Bio-Rad iQ5 Sequence Detection System (Bio-Rad, Hercules,
CA, USA). The PCR conditions were as follows: one cycle at 95°C for
10 min followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min.
The results were normalized to those obtained with GAPDH. Each
sample was assayed in triplicate and the experiment was repeated
twice. The CT value was measured and 2−ΔCT (ΔCT = CT -
CTGAPDH) was defined as the quantity of the amplified
fragment. The primer sequences are listed in Table I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| GAPDH |
CCATGAGAAGTATGACAACAGCC |
GGGTGCTAAGCAGTTGGTG |
| VE-cadherin |
GACGCCCGGCCTTCCCTCTA |
TCGTGGTCCGCCTCGTCCTT |
| VEGF |
TTACGGTCTGTGTCCAGTGTA |
TTCTCTGTTATGTTGCCAGCC |
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical analyses were performed using SPSS 11.0 statistical
software (SPSS, Inc., Chicago, IL, USA). Statistical significance
was analyzed by one-way analysis of variance. If significance was
observed, the Dunnett’s post-hoc test was used to determine the
difference between treatment groups and the untreated group.
P<0.05 was considered to indicate a statistically significant
difference.
Results
RSG inhibits the migration and invasion
of PC-3 cells
VM is associated with cell migration and invasion.
To examine the effect of RSG on PC-3 cells, an in vitro
wound healing assay was performed. Since TZDs activate PPARα and
PPARδ receptors at concentrations >10 μM (21), TZDs were used at concentrations of
10 μM or less. Following incubation with different concentrations
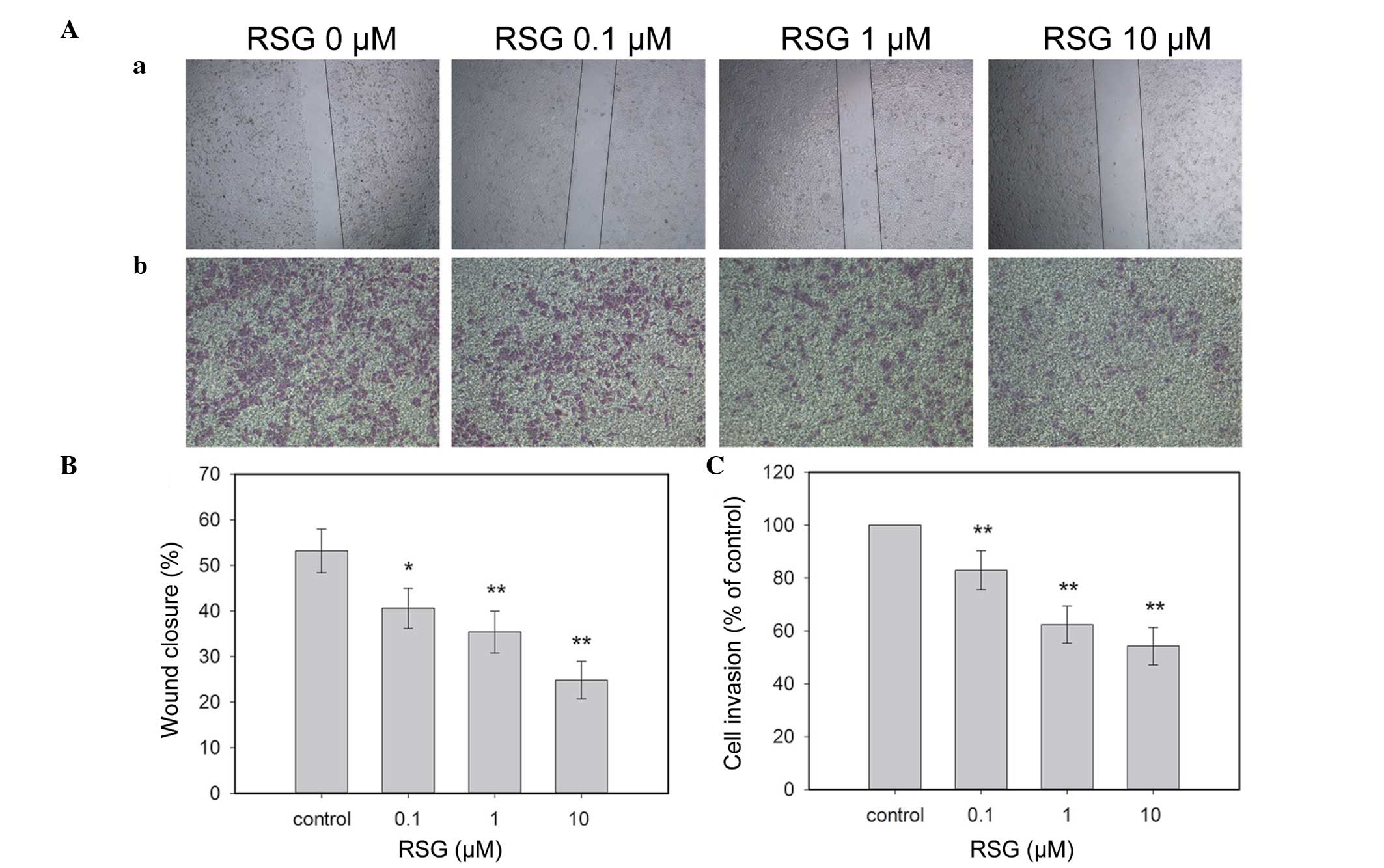
of RSG for 24 h, the migration of PC-3 cells to the denuded zone
was suppressed in a dose-dependent manner (Fig. 1Aa and 1B). These results revealed
that RSG significantly inhibited the motility of PC-3 cells.
Using a chamber invasion assay, it was revealed that
RSG suppressed the invasion of PC-3 cells across the
Matrigel-coated filter in a dose-dependent manner. Treatment with
0.1, 1 and 10 μM of RSG inhibited 17.01, 27.62 and 35.74% of cell
invasion, respectively (Fig. 1Ab and
1C). The results indicated that RSG markedly inhibited the
invasion of PC-3 cells.
RSG prevents the development of VM in a
PPARγ-dependent manner
In order to elucidate the VM capability of PC-3
cells, a well-established in vitro model of VM formation was
utilized. PC-3 cells were able to form patterned matrix VM or
typical pipe-like VM networks within the Matrigel medium within 48
h.
Next, the present study investigated the effect of
RSG on VM formed by PC-3 cells. The in vitro tube formation
assay demonstrated that RSG effectively inhibited the formation of
capillary-like structures in a dose-dependent manner (Fig. 2A and B). Western blot analysis of
whole-cell lysates from these experimental samples was conducted to
assess the expression of VM-associated markers, including
VE-cadherin. The results demonstrated that RSG significantly
decreased the expression of VE-cadherin at the mRNA and protein
levels (P<0.05; Fig. 2C and
D).
Although RSG was able to affect the VM process and
the expression of VE-cadherin, the mechanisms underlying these
events remain to be elucidated. To confirm whether these effects
were dependent upon the activation of PPARγ, GW9662, a PPARγ
antagonist, was used to inhibit the function of PPARγ in prostate
cancer cells. The results demonstrated that GW9662 inhibited
RSG-induced reduction of VM formation. Furthermore, the RSG-induced
downregulation of VE-cadherin in PC-3 cells was attenuated upon
addition of GW9662 (Fig. 2). Taken
together, these data suggested that RSG inhibited the VM formation
of PC-3 cells in a PPARγ-dependent manner.
Effect of RSG on the expression of
VEGF
Since prostate cancer cells induce angiogenesis
through the expression of angiogenic factors, including VEGF, the
present study evaluated whether VEGF stimulates the formation of VM
in PC-3 cells, utilizing an in vitro tube formation assay.
Quantification of the tubule number demonstrated that rhVEGF165
effectively promoted the formation of capillary-like structures
(Fig. 5A and B). Furthermore, RSG
inhibited the expression of VEGF in PC-3 cells. The secretion of
VEGF protein was reduced to 59.38, 39.64 and 29.99% in PC-3 cells
in the presence of RSG as compared with the control (Fig. 3A). Similar results were observed
while detecting the VEGF mRNA levels by qPCR (Fig. 3B). Furthermore, the RSG-induced
downregulation of VEGF in PC-3 cells was attenuated upon addition
of GW9662 (Fig. 3). The present
study also demonstrated that the addition of rhVEGF165 partly
inhibited RSG-induced downregulation of VM formation and
VE-cadherin expression (Fig. 5A and
C). These data suggest that the inhibition VM of PC-3 cells by
RSG may partly occur through suppressing VEGF expression.
RSG modulates the expression of VEGF and
VM via the AKT pathway
Several studies have indicated that signaling
proteins, including PI3K and AKT, are involved in the regulation of
the expression of MMPs and the promotion of metastasis (22). To understand the signaling pathways
responsible for VEGF production and VM formation, the activation of
AKT was examined. As expected, RSG downregulated the
phosphorylation of AKT, the active form of PI3K/AKT, in a dose- and
time-dependent manner (Fig. 4),
which indicated that AKT may also be involved in the inhibition of
VM formation in PC-3 cells.
To further investigate whether the inhibition of VM
formation and VE-cadherin expression proceeded through the
inhibition of the PI3K-AKT signaling pathway, PC-3 cells were
treated with a PI3K inhibitor (LY294002; 20 μM) for 48 h. This
disturbance of the AKT pathway resulted in the downregulation of
VEGF secretion in PC-3 cells (Fig.
5D), which is consistent with the results of other studies
(23). In addition, treatment with
LY294002 significantly reduced cell VM formation and VE-cadherin
expression (Fig. 5A and C),
suggesting that the inhibition of VM formation and VEGF expression
by RSG partly occurred through suppressing the PI3K pathway. Of
note, the addition of rhVEGF165 eradicated the inhibitory effect of
RSG on the phosphorylation of AKT (Fig. 4A). These data suggest that the
inactivation of the PI3K/AKT pathway by RSG may be mediated by the
inhibition of VEGF in an autocrine manner.
Discussion
Globally, 903,500 new cases of prostate cancer were
estimated to occur, second only to lung cancer in males, and
258,400 mortalities from prostate cancer were estimated for 2011
(24). The majority of patients
with localized early stage disease do not develop metastases and do
not succumb to prostate cancer. However, among patients with
castration-resistant metastatic prostate cancer, >80% of
patients develop bone metastasis, which is the most common site of
metastases in this group (25).
Therefore, understanding the molecular mechanisms of prostate
cancer invasiveness and angiogenesis are of significant value to
the prostate cancer field.
Targeting the blood supply by inhibiting the
formation of blood vessels may lead to tumor growth arrest.
Numerous angiogenesis inhibitors have been therapeutically used in
preclinical and clinical settings (26). VEGF receptor tyrosine kinase
inhibitors and a VEGF-neutralizing antibodies have been clinically
validated to target VEGF or its receptors as an anticancer
treatment. However, amongst other limitations of angiogenesis
inhibitors, tubule formation was not inhibited. For instance,
treatment with angiogenesis inhibitors did not inhibit tube
formation of aggressive uveal and cutaneous melanoma cells in
vitro (27). Of note, recent
findings indicated that anti-angiogenesis treatment may elicit the
malignant progression of various types of tumor (28). Furthermore, the inhibition of
endothelial angiogenesis may unintentionally promote VM formation
in tumors (29), as recently
evidenced by a study demonstrating increased VM channels in
tumor-bearing mice receiving short-term treatment of the anti-VEGF
monoclonal antibody bevacizumab (30). It has been demonstrated that
several drugs were able to inhibit VM (31,32).
However, at present, data on the effect of anti-angiogenesis
treatment on VM network formation remain inconsistent. The
inhibition of endothelial cell migration by PPARγ ligands has been
described, bolstering the anti-angiogenic activity of PPAR ligands
(17,33). An earlier study has demonstrated
that the activation of PPARγ by TZDs inhibits angiogenesis and
neovascularization in vitro and in vivo and inhibits
the release of VEGF from smooth muscle cells. The present study
revealed that RSG was able to inhibit VM formation of the highly
aggressive prostate cancer cell line PC-3 in vitro in a
dose- and PPARγ-dependent manner. The results demonstrated that RSG
significantly decreased the expression of VM-associated markers,
including VE-cadherin, at the mRNA and protein levels, in line with
the results of functional studies. RSG also reduced the migration
and invasion capacity of PC-3 cells. Therefore, PPARγ agonists may
be further used to unravel new biological mechanisms that drive VM,
angiogenesis and tumor progression.
VEGF and its receptors appear to contribute to VM
formation in certain types of cancer (10,34).
However, VEGF did not demonstrate any effect on VM formation of
Ewing sarcoma cells and hepatocellular carcinoma cell lines
(35,36), and the effect of VEGF on the VM
formation of prostate cancer cells remains to be elucidated. The
present study demonstrated that rhVEGF165 increased the VM
formation of PC-3 cells. Notably, it was demonstrated that VEGF was
downregulated at the mRNA and protein secretion levels upon RSG
treatment. These effects were dose dependent and the concentrations
used in the present study were consistent with those reported by
others (17). However, another
study reported that TZDs enhanced VEGF expression in non-small cell
lung cancer cells (37). The
variation of outcome from those studies may be due to differences
in culture and experimental conditions, the type and site of
carcinoma or a combination of these factors.
Besides the functional study, elucidation of the
mechanisms is crucial for understanding RSG as a potential
anticancer agent. PI3K and AKT are important for the signaling of
angiogenic growth factors that are closely associated with cancer
cell migration, proliferation, angiogenesis and VM (38,39).
Although LY294002 is useful for conducting preclinical studies on
PI3K inhibition, its poor solubility and narrow therapeutic index
preclude its use in humans. It has been reported that the PPARγ
agonist was able to decrease the phosphorylation of AKT (33,40).
The present study used the PPARγ agonist, RSG, to stimulate PC-3
cells. Our results demonstrated that RSG downregulated the
phosphorylation of AKT in a dose- and time-dependent manner. In
order to examine this possibility, the PI3K inhibitor LY294002 was
used to inhibit these pathways to establish a link with VM
formation. The data demonstrated that the disturbance of the AKT
pathway resulted in the inhibition of the formation of VM in PC-3
cells and the downregulation of VE-cadherin and VEGF expression in
PC-3 cells. Therefore, the present study suggested that RSG
inhibited VM formation of PC-3 cells, which may be partly through
suppressing PI3K-associated pathways. In addition, the stimulation
of the cells with rhVEGF165 in the culture medium attenuated the
inhibitory effect of RSG on the AKT pathway and cancer phenotypes.
Previous studies demonstrated that the PI3K/AKT pathway may be
activated by growth factors or hypoxia and are able to promote
tumor angiogenesis through the enhanced expression of
hypoxia-inducible factor 1 and VEGF (41). In addition, in the present study
and in other studies (42,43), suppression of these pathways by a
PI3K inhibitor markedly reduced VEGF protein levels. Thus, the
present study concluded that the PI3K/AKT pathway and VEGF may
generate a positive autocrine loop. These findings associated with
the dual inhibition of the VEGF/PI3K/AKT cascade and the
PI3K/AKT/VEGF cascade by RSG treatment in prostate cancer cells
provided insights into the molecular mechanisms of the anti-tumor
effects of RSG and support its potential clinical application.
In conclusion, the present study indicated that RSG
effectively inhibited VM formation of PC-3 cells in vitro by
inhibiting VEGF gene expression and phosphorylation of AKT. The
present study may provide preliminary evidence for further
elucidating the mechanisms underlying VM inhibition by TZDs and its
mode of action. RSG was used as a proof-of-principle to determine
the efficacy of PPARγ agonists in the inhibition of VM
formation.
Acknowledgements
This study was supported by a grant from the Health
Department of Hubei Province’s Program for Youth Science and
Technology Talent (QJX2010-5) and the Key Laboratory of Cancer
Invasion and Metastasis of the Ministry of Education of People’s
Republic of China (no. 200804871-051).
References
|
1
|
Hanahan D and Folkman J: Patterns and
emerging mechanisms of the angiogenic switch during tumorigenesis.
Cell. 86:353–364. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Döme B, Hendrix MJ, Paku S, Tóvári J and
Tímár J: Alternative vascularization mechanisms in cancer:
Pathology and therapeutic implications. Am J Pathol. 170:1–15.
2007.PubMed/NCBI
|
|
3
|
Maniotis AJ, Folberg R, Hess A, et al:
Vascular channel formation by human melanoma cells in vivo and in
vitro: vasculogenic mimicry. Am J Pathol. 155:739–752. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Basu GD, Pathangey LB, Tinder TL, Gendler
SJ and Mukherjee P: Mechanisms underlying the growth inhibitory
effects of the cyclo-oxygenase-2 inhibitor celecoxib in human
breast cancer cells. Breast Cancer Res. 7:R422–R435. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
El Hallani S, Boisselier B, Peglion F, et
al: A new alternative mechanism in glioblastoma vascularization:
tubular vasculogenic mimicry. Brain. 133:973–982. 2010.PubMed/NCBI
|
|
6
|
Sharma N, Seftor RE, Seftor EA, et al:
Prostatic tumor cell plasticity involves cooperative interactions
of distinct phenotypic subpopulations: role in vasculogenic
mimicry. Prostate. 50:189–201. 2002. View Article : Google Scholar
|
|
7
|
Sun B, Zhang D, Zhang S, Zhang W, Guo H
and Zhao X: Hypoxia influences vasculogenic mimicry channel
formation and tumor invasion-related protein expression in
melanoma. Cancer Lett. 249:188–197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Millimaggi D, Mari M, D’ Ascenzo S, Giusti
I, Pavan A and Dolo V: Vasculogenic mimicry of human ovarian cancer
cells: role of CD147. Int J Oncol. 35:1423–1428. 2009.PubMed/NCBI
|
|
9
|
Zhang S, Zhang D and Sun B: Vasculogenic
mimicry: current status and future prospects. Cancer Lett.
254:157–164. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang JY, Sun T, Zhao XL, et al: Functional
significance of VEGF-a in human ovarian carcinoma: role in
vasculogenic mimicry. Cancer Biol Ther. 7:758–766. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu R, Yang K, Meng C, Zhang Z and Xu Y:
Vasculogenic mimicry is a marker of poor prognosis in prostate
cancer. Cancer Biol Ther. 13:527–533. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kota BP, Huang TH and Roufogalis BD: An
overview on biological mechanisms of PPARs. Pharmacol Res.
51:85–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim S, Lee JJ and Heo DS: PPARgamma
ligands induce growth inhibition and apoptosis through p63 and p73
in human ovarian cancer cells. Biochem Biophys Res Commun.
406:389–395. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lyles BE, Akinyeke TO, Moss PE and Stewart
LV: Thiazolidinediones regulate expression of cell cycle proteins
in human prostate cancer cells via PPARgamma-dependent and
PPARgamma-independent pathways. Cell Cycle. 8:268–277. 2009.
View Article : Google Scholar
|
|
15
|
Fujita M, Yagami T, Fujio M, et al:
Cytotoxicity of troglitazone through PPARgamma-independent pathway
and p38 MAPK pathway in renal cell carcinoma. Cancer Lett.
312:219–227. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim KY, Ahn JH and Cheon HG:
Anti-angiogenic action of PPARgamma ligand in human umbilical vein
endothelial cells is mediated by PTEN upregulation and VEGFR-2
downregulation. Mol Cell Biochem. 358:375–385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Panigrahy D, Singer S, Shen LQ, et al:
PPARgamma ligands inhibit primary tumor growth and metastasis by
inhibiting angiogenesis. J Clin Invest. 110:923–932. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tian L, Zhou J, Casimiro MC, et al:
Activating peroxisome proliferator-activated receptor gamma mutant
promotes tumor growth in vivo by enhancing angiogenesis. Cancer
Res. 69:9236–9244. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lissitzky JC, Parriaux D, Ristorcelli E,
Verine A, Lombardo D and Verrando P: Cyclic AMP signaling as a
mediator of vasculogenic mimicry in aggressive human melanoma cells
in vitro. Cancer Res. 69:802–809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu H, Chen A, Guo F and Yuan L: A
short-hairpin RNA targeting osteopontin downregulates MMP-2 and
MMP-9 expressions in prostate cancer PC-3 cells. Cancer Lett.
295:27–37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamashita D, Shimizu M and Osumi T:
Mechanism for the action of PPARs. Nihon Rinsho. 63:536–537.
2005.(In Japanese).
|
|
22
|
Shukla S, Maclennan GT, Hartman DJ, Fu P,
Resnick MI and Gupta S: Activation of PI3K-Akt signaling pathway
promotes prostate cancer cell invasion. Int J Cancer.
121:1424–1432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang J, Wang J, Sun Y, Song W, Nor JE,
Wang CY and Taichman RS: Diverse signaling pathways through the
SDF-1/CXCR4 chemokine axis in prostate cancer cell lines leads to
altered patterns of cytokine secretion and angiogenesis. Cell
Signal. 17:1578–1592. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
25
|
de Bono JS, Oudard S, Ozguroglu M, et al:
Prednisone plus cabazitaxel or mitoxantrone for metastatic
castration-resistant prostate cancer progressing after docetaxel
treatment: a randomised open-label trial. Lancet. 376:1147–1154.
2010.
|
|
26
|
Samaranayake H, Määttä AM, Pikkarainen J
and Ylä-Herttuala S: Future prospects and challenges of
antiangiogenic cancer gene therapy. Hum Gene Ther. 21:381–396.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van der Schaft DW, Seftor RE, Seftor EA,
et al: Effects of angiogenesis inhibitors on vascular network
formation by human endothelial and melanoma cells. J Natl Cancer
Inst. 96:1473–1477. 2004.PubMed/NCBI
|
|
28
|
Pàez-Ribes M, Allen E, Hudock J, et al:
Antiangiogenic therapy elicits malignant progression of tumors to
increased local invasion and distant metastasis. Cancer Cell.
15:220–231. 2009.PubMed/NCBI
|
|
29
|
Qu B, Guo L, Ma J and Lv Y:
Antiangiogenesis therapy might have the unintended effect of
promoting tumor metastasis by increasing an alternative circulatory
system. Med Hypotheses. 74:360–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu Y, Li Q, Li XY, Yang QY, Xu WW and Liu
GL: Short-term anti-vascular endothelial growth factor treatment
elicits vasculogenic mimicry formation of tumors to accelerate
metastasis. J Exp Clin Cancer Res. 31:162012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Itzhaki O, Greenberg E, Shalmon B, et al:
Nicotinamide inhibits vasculogenic mimicry, an alternative
vascularization pathway observed in highly aggressive melanoma.
PLoS One. 8:e571602013. View Article : Google Scholar
|
|
32
|
Fan YZ and Sun W: Molecular regulation of
vasculogenic mimicry in tumors and potential tumor-target therapy.
World J Gastrointest Surg. 2:117–127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Goetze S, Eilers F, Bungenstock A, et al:
PPAR activators inhibit endothelial cell migration by targeting
Akt. Biochem Biophys Res Commun. 293:1431–1437. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vartanian A, Stepanova E, Grigorieva I,
Solomko E, Baryshnikov A and Lichinitser M: VEGFR1 and PKCalpha
signaling control melanoma vasculogenic mimicry in a VEGFR2
kinase-independent manner. Melanoma Res. 21:91–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van der Schaft DW, Hillen F, Pauwels P, et
al: Tumor cell plasticity in Ewing sarcoma, an alternative
circulatory system stimulated by hypoxia. Cancer Res.
65:11520–11528. 2005.PubMed/NCBI
|
|
36
|
Lirdprapamongkol K, Chiablaem K, Sila-Asna
M, Surarit R, Bunyaratvej A and Svasti J: Exploring stemness gene
expression and vasculogenic mimicry capacity in well- and
poorly-differentiated hepatocellular carcinoma cell lines. Biochem
Biophys Res Commun. 422:429–435. 2012. View Article : Google Scholar
|
|
37
|
Yoshizaki T, Motomura W, Tanno S, Kumei S,
Yoshizaki Y and Okumura T: Thiazolidinediones enhance vascular
endothelial growth factor expression and induce cell growth
inhibition in non-small-cell lung cancer cells. J Exp Clin Cancer
Res. 29:222010. View Article : Google Scholar
|
|
38
|
Hess AR, Seftor EA, Seftor RE and Hendrix
MJ: Phosphoinositide 3-kinase regulates membrane Type 1-matrix
metalloproteinase (MMP) and MMP-2 activity during melanoma cell
vasculogenic mimicry. Cancer Res. 63:4757–4762. 2003.PubMed/NCBI
|
|
39
|
Chetty C, Lakka SS, Bhoopathi P and Rao
JS: MMP-2 alters VEGF expression via alphaVbeta3 integrin-mediated
PI3K/AKT signaling in A549 lung cancer cells. Int J Cancer.
127:1081–1095. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen WC, Lin MS and Bai X: Induction of
apoptosis in colorectal cancer cells by peroxisome
proliferators-activated receptor gamma activation up-regulating
PTEN and inhibiting PI3K activity. Chin Med J (Engl).
118:1477–1481. 2005.PubMed/NCBI
|
|
41
|
Trisciuoglio D, Iervolino A, Zupi G and
Del Bufalo D: Involvement of PI3K and MAPK signaling in
bcl-2-induced vascular endothelial growth factor expression in
melanoma cells. Mol Biol Cell. 16:4153–4162. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pore N, Gupta AK, Cerniglia GJ and Maity
A: HIV protease inhibitors decrease VEGF/HIF-1alpha expression and
angiogenesis in glioblastoma cells. Neoplasia. 8:889–895. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhong H, Chiles K, Feldser D, et al:
Modulation of hypoxia-inducible factor 1alpha expression by the
epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP
pathway in human prostate cancer cells: implications for tumor
angiogenesis and therapeutics. Cancer Res. 60:1541–1545. 2000.
|