Introduction
Liver cancer is one of the most life-threatening
diseases in the world, and encompasses several histologically
different primary hepatic malignancies, including
cholagiocarcinoma, hepatoblastoma and hemangiosarcoma. However, the
most common type of liver cancer is hepatocellular carcinoma (HCC)
which accounted for 70–85% of all cases in 2011 (1). Liver carcinogenesis is a multistep
process. The presence of specific risk factors promotes gene
damage, leading to a cascade of molecular and cellular deregulation
that ultimately results in transformation of hepatocytes (2). The ideal therapy for HCC is surgical
resection and transplantation, but a lack of liver donors limits
the use of these methods. Thus far, transarterial chemoembolization
(TACE) is the preferred treatment choice for liver cancer and
improves the survival rate of patients (3). Regardless of extensive research into
procedures, devices and the application of anti-cancer agents for
TACE, the absence of a suitable animal model to replace the rodent
model has been the major factor impeding progression in this
field.
Several genetically engineered models of HCC have
been developed since the early 1980s when the transgenic mouse
technique was first introduced (4). Transgenic mice expressing Simian
virus 40 large T-antigen in their hepatocytes developed HCC at the
age of 8 months (5). In another
study, all transforming growth factor α (TGF-α) and c-Myc
(Myc) transgenic male mice presented HCC within 8 months (6). Establishing successful animal models
of HCC is crucial for basic and translational studies of HCC. A
plethora of HCC mouse models are currently available, which has
provided researchers with the opportunities to assess tumor-host
interactions, perform drug screenings, mimic the complex multistep
process of liver carcinogenesis and conduct various therapeutic
experiments (2). However, no mouse
model is ideal for the purpose of studying surgical procedures and
devices, due to their small body size relative to humans. Thus,
alternative models are required to overcome this size
limitation.
One of the best candidates is the pig model, as pigs
possess anatomical and physiological characteristics similar to
those of humans (7). Studies have
previously been conducted in pig models of human diseases using
transgenic and somatic cell nuclear transfer (SCNT) technologies
(8,9). In the present study, the
hepatocyte-specific TGF-α and Myc overexpression strategy of
previous mouse models (4,6) was adopted in order to allow for the
generation of a pig model of HCC in future. To minimize off-target
expression of the two proto-oncogenes in the present study,
progressive in vitro experiments were performed using a
vector constructed in liver and kidney cell lines. The resulting
transgenic cell lines were obtained and the insertion of the
transgenes in their genomic DNA was checked to confirm that they
could be used as donor cells for SCNT.
Materials and methods
Cell culture
Unless otherwise indicated, all cells were grown at
37°C in 5% CO2, and all cell culture materials were
obtained from PAA Laboratories GmbH (Pasching, Austria). Hep G2 and
293T cells were provided by Dr. Young-Wook Cho (Korean Basic
Science Institute, Chuncheon, Korea). To obtain porcine
fibroblasts, a pig uterus containing a fetus (male, ~30 embryonic
days old) was transported to the laboratory from a local
slaughterhouse (Wonju, Korea). After sterilizing, fetal ears and
skin were isolated and minced with a surgical blade in a culture
dish (35- or 100-mm; SPL Life Science Co., Gyeonggi-do, Republic of
Korea), and subjected to Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 0.1% (w/v) trypsin/1 mM EDTA for 1–2 h.
Trypsinized cells were subsequently cultured for 6–8 days in DMEM
supplemented with 10% (v/v) FBS and a 10 μg/ml
penicillin/streptomycin solution. When the cells were fully
confluent, they were collected by trypsinization and were frozen in
DMEM supplemented with 40% FBS and 10% dimethyl sulfoxide. Porcine
peripheral blood monocytes were isolated using lymphocyte
separation medium (LSM; Sigma-Aldrich, St. Louis, MO, USA) from
freshly drawn peripheral venous blood obtained from domestic pigs.
Briefly, EDTA (1.5 mg per ml of blood; Sigma-Aldrich) treated blood
(10 ml) was diluted with equal volumes of PBS and 10 ml of LSM was
carefully poured into a centrifuge tube (SPL Life Science Co.). The
tube was centrifuged at 500 × g at room temperature for 30 min to
create a blood-LSM interphase. The mononuclear cell layer was
collected into a new tube and diluted with three volumes of PBS.
The tube was centrifuged at 500 × g at room temperature for 10 min.
To eliminate RBC contamination, the cell pellet was further treated
with RBC lysis buffer (Intron Biotechnology, Seongnam-si, Korea).
All cells were cultured in Dulbecco’s modified Eagle’s medium
(DMEM) containing 10% fetal bovine serum (FBS), 50 U/ml penicillin
and 5 μg/ml streptomycin.
RNA extraction and genomic DNA
extraction
Hep G2 cells and peripheral blood mononuclear cells
from the pig were subjected to TRIzol reagent (Invitrogen,
Carlsbad, CA, USA) to extract total RNA. The RNA concentrations
were determined with a spectrophotometer (NanoDrop 2000c; Thermo
Scientific, Wilmington, DE, USA) and the RNA was
reverse-transcribed into first-strand complementary DNA using
Moloney Murine Leukemia Virus Reverse Transcriptase (Invitrogen)
according to the manufacturer’s instructions. Genomic DNA was
isolated by a G-DEX™ IIc Genomic DNA Extraction kit (Intron
Biotechnology).
Polymerase chain reaction (PCR) and
quantitative (q)PCR
PCR reactions were performed with LA-Taq polymerase
(Takara Bio, Inc., Otsu, Japan) or i-StarTaq polymerase (Intron
Biotechnology). The name of PCR fragments, the sequences of
primers, the sources of genes (GenBank ID) and the added
restriction enzymes are described in Table I. Primers were synthesized by
Macrogen (Seoul, Korea). The PCR reactions involved denaturing at
95°C for 30 sec, annealing at 60°C for 30 sec, and extension at
72°C for 30 sec to 3 min depending on the size of the products (1
min/kb). The PCR products were subjected to cloning processes
and/or separated on an agarose gel (1 or 2%; Invitrogen), stained
with ethidium bromide (Invitrogen) and photographed under UV
illumination (AE-9150 EZ-Capture II; Atto Corporation, Tokyo,
Japan). For gene quantification, qPCR was performed using FG Power
SYBR-Green PCR Master mix (Applied Biosystems, Carlsbad, CA, USA)
and Eco Real-Time PCR system (Illumina, San Diego, CA, USA).
 | Table IPrimer details. |
Table I
Primer details.
| Name | GenBank ID | Direction | Restriction
enzyme | Sequence (5′ to
3′) |
|---|
| Myc cDNA | NM_001005154 | F | SalI |
TGGACGCTGGATTTCCTTCGGATA |
| | R | BamHI |
TTATGGGCAAGAGTTCCGTAGCTG |
| TGF-α cDNA | NM_214251 | F | EcoRI |
CGTAAAATGGTCCCCTCGGCTGGA |
| | R | BamHI |
TCAGACCACTGTTTCTGAGTGGCA |
| Porcine albumin
promoter (−3,044 nt) | NC_010450 | F | NheI |
TCTCTTCTAAATGATCAGCATATA |
| Porcine albumin
promoter (−2,033 nt) | | F | NheI |
TTGCAGTGCAAAGTAGGTGGAGAG |
| Porcine albumin
promoter (−1,036 nt) | | F | NheI |
TCAGAATTTGGGGTGGGAAAAGTA |
| Porcine albumin
promoter (−7 nt) | | R | HindIII |
AAAGGCTTGTGGGGTTGATA |
| Human albumin
mRNA | NM_000477 | F | |
ACTTTTATGCCCCGGAACTC |
| | R | |
TGGAGACTGGCACACTTGAG |
| Human actin
mRNA | NM_001101 | F | |
GGACTTCGAGCAAGAGATGG |
| | R | |
AGCACTGTGTTGGCGTACAG |
| Confirming
primer-1 | NM_001005154 | F | |
GGAAGAGGCGAGAACAGTTG |
| Confirming
primer-2 | NM_214251 | F | |
TGATACACTGCTGCCAGGTC |
| Confirming
primer-3 | IRES | R | |
GAGGAACTGCTTCCTTCACG |
| Confirming
primer-4 | NC_010450 | F | |
TGCTTATTCCAGGGGTGTGT |
| Confirming
primer-5 | NC_010450 | R | |
AAGCTCCTTCATGTGCAAAA |
Vector construction
Unless otherwise indicated, all restriction enzymes
and cloning enzymes were obtained from Enzynomics (Daejeon, Korea).
The genes isolated by PCR were cloned into a yT&A vector (TA
vector; RBC Bioscience Corp., Taipei, Taiwan), and the insertion of
nucleotide sequences (Macrogen) was confirmed and/or subjected to
further sub-cloning. To create the overexpression vector, two
proto-oncogenes were inserted into pIRES2-DsRed-Express™ (Clontech
Laboratories, Mountain View, CA, USA). For the luciferase assay,
several promoter regions of the porcine albumin (Alb) gene were
inserted in pGL4.10 (Promega Corporation, Madison, WI, USA). The
selected promoter region was further ligated upstream of the
proto-oncogenes of the overexpression vector. The two
overexpression vectors, pAlb-TGF-RFP and pAlb-Myc-RFP, were
displayed by schematic diagrams using PlasmTM (version 2.1.5.30;
http://biofreesoftware.com).
Fluorescence signals were observed using a fluorescence microscope
(Nikon, Tokyo, Japan).
Transfection and promoter assessment
Transfection was conducted using Lipofectamine™ 2000
(Invitrogen) according to the manufacturer’s instructions. 293T
cells and Hep G2 cells (3×105 cells) were plated in
24-well plates (SPL Life Sciences) one day prior to transfection.
The promoter constructs (1.6 μg) and pRL-TK (3.2 ng; Promega
Corporation) were mixed with serum-free DMEM containing
Lipofectamine™ 2000 (Invitrogen) and then added to the wells.
Following incubation overnight, the cell culture media were
replaced with DMEM containing 10% FBS with 10 μg/ml
penicillin/streptomycin solution and incubated for an additional
night. Cellular lysates were assayed for luciferase activity using
the Dual-Luciferase Reporter Assay system and a GloMax 20/20
Luminometer (Promega Corporation). The relative luciferase activity
(%) was calculated as luciferase activity/Renilla luciferase
activity.
Western blotting
The cell lysates were prepared using mild lysis
buffer (150 mM NaCl, 1% Triton X-100, 50 mM Tris-base, pH 8.0;
Sigma-Aldrich). Western blots were prepared, probed at 4°C
overnight with c-Myc antibody (SC-40), TGF-α antibody
(SC-36) or actin antibody (SC-1615), and then further probed at
room temperature for 3 h with the horseradish peroxidase-conjugated
secondary antibodies (SC-2005 or SC2020; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA). The membranes were visualized using
PowerOpti-ECL solution (BioNote, Hwasung, Korea) as described in
the manufacturer’s instructions and images were captured using the
EZ-Capture II (Atto Corporation).
Establishment of transgenic cell
lines
To determine an appropriate G-418 (Roche,
Indianapolis, IN, USA) concentration, 500 porcine fetal fibroblasts
were seeded in 96-well plates (SPL Life Sciences) and were treated
with various dosages (15.7, 31.1, 62.3, 125, 250, 500, 1,000 and
2,000 μg/ml) of G-418 or triton X-100 (0.01%; Sigma-Aldrich) for 3
days. The survival rate was measured using Cell Counting kit-8™
(Dojindo, Kumamoto, Japan). To create stable cell lines, porcine
fetal fibroblasts (5×106 cells) were plated in a 100-mm
culture dish (SPL Life Sciences) 1 day prior to transfection, and
transfected with 24 μg linearized overexpression vectors
using Lipofectamine™ 2000 (Invitrogen). Following incubation for 6
h, the media were replaced with DMEM containing 10% FBS and 125
μg/ml G-418 for 24 h. The concentration of G-418 was
gradually reduced to 30 μg/ml at which point colony
formation occurred and then the colony was transported to a 6-well
plate (SPL Life Sciences) in order to increase the cell number for
further study. Fully confluent colonies in the 6-well plate were
divided into two portions and subjected to PCR-based genotyping or
stored (5×105 cells per vial) in the liquid nitrogen
tank until required for SCNT in future studies.
Data analysis and ethics
A statistical analysis was performed using GraphPad
Prism (version 5 for Windows, GraphPad Software, San Diego, CA,
USA). The present study was approved by the ethics committee of
Kangwon National University (Chuncheon, Republic of Korea).
Results
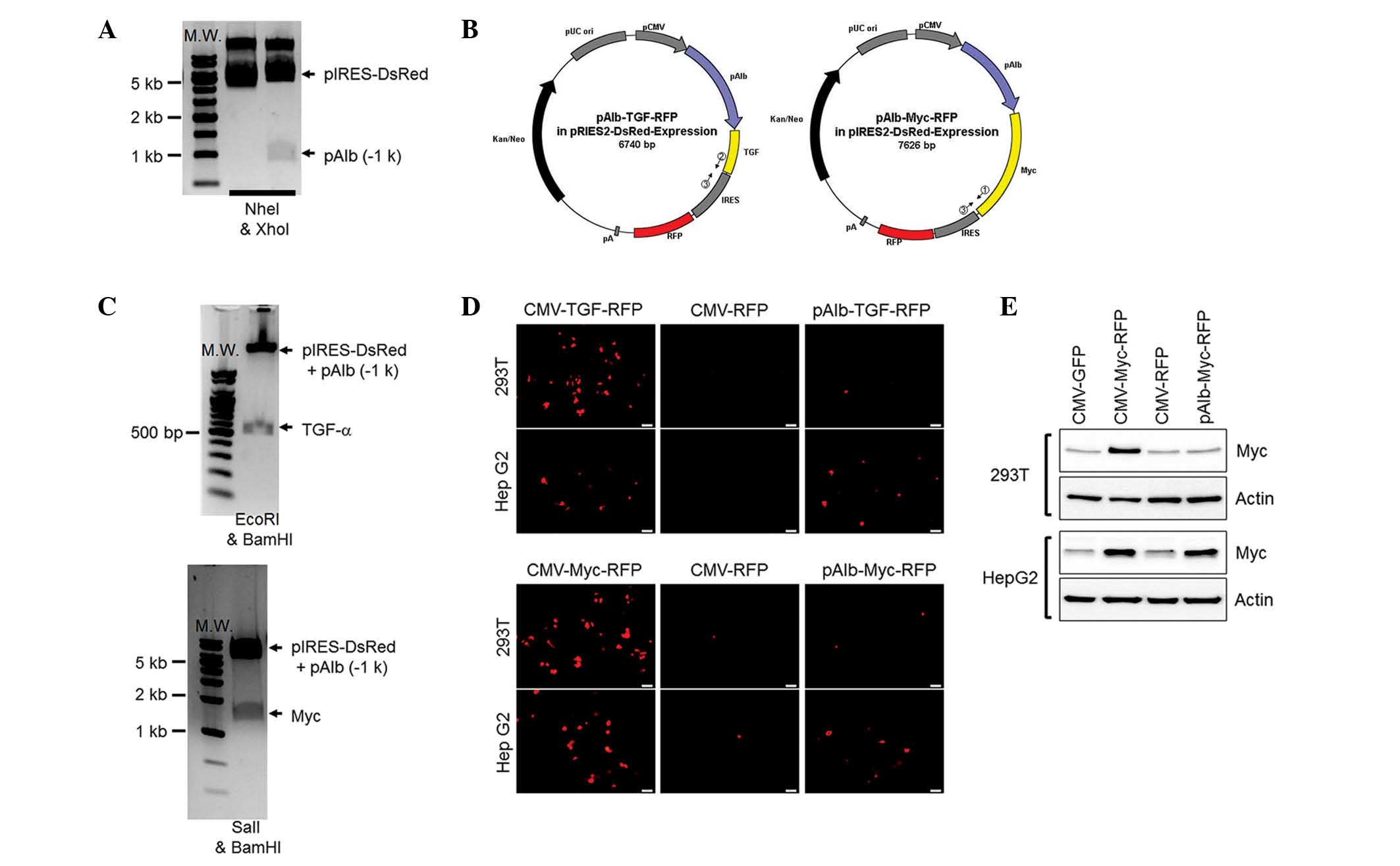
Cloning of porcine TGF-α and Myc
genes
To create a mechanism to induce carcinogenesis in
the pig liver, two proto-oncogenes (TGF-α and Myc) were isolated
from the porcine peripheral blood monocytes by reverse
transcription-PCR (Fig. 1A). The
isolated genes were cloned into a TA vector (Fig. 1B) and the nucleotide sequences were
confirmed by a sequencing analysis (data not shown). TGF-α or Myc
genes were further cloned into the overexpression vector containing
the gene for red fluorescent protein (RFP; the marker of
translation) conjugated by the internal ribosomal entry site
(Fig. 1C). The translational
activities were further checked in a cell line by transient
transfection of these overexpression vectors controlled by a
universal (cytomegalovirus; CMV) promoter. The vectors presented
RFP signals, which indicated the successful translation of TGF-α or
Myc in vivo (Fig. 1D). In
addition, the overexpressed TGF-α and Myc proteins in porcine
fibroblasts were confirmed by immunoblotting (Fig. 1E). Thus, two proto-oncogenes were
isolated from the pig to induce carcinogenesis in future
studies.
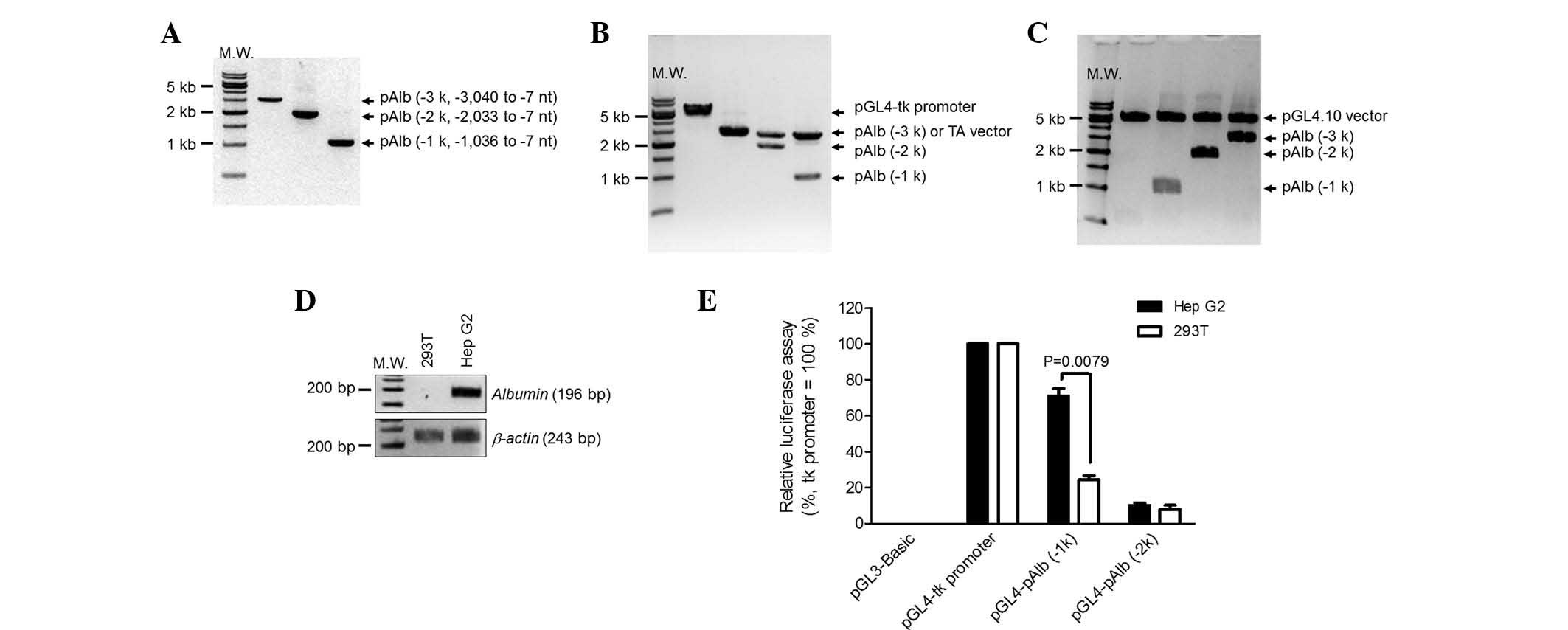
Assessment of porcine Alb gene as a
promoter
To selectively induce the proto-oncogenes in
hepatocytes, the liver-specific promoter Alb (pAlb) was assessed.
Several regions of the porcine Alb promoter were isolated from the
genomic DNA of fetal fibroblasts using PCR (Fig. 2A) and then cloned into the TA
vector (Fig. 2B). The isolated
promoters were sub-cloned into the vector encoding luciferase to
assay the relative promoter activity (Fig. 2C). To confirm a hepatocyte-specific
Alb promoter, kidney (293T) and liver (Hep G2) cell lines were
used. As expected, albumin mRNA was detected in the Hep G2 cells,
but not in the 293T cells (Fig.
2D). The luciferase vectors were transiently transfected into
the two cell lines and luciferase enzyme activity was measured
(Fig. 2E). pAlb (−1k) represented
70% of promoter activity in Hep G2 cells when compared with a
thymidine kinase (tk) promoter, which was used as a positive
control. The same promoter in 293T cells represented <20% of the
activity. However, the luciferase activities of pAlb (−2k) were
lower than that of pAlb (−1k) and not different between cell types.
The longest promoter regions, pAlb (−3k), presented very weak
promoter activities similar to a negative control, pGL3-Basic. On
this account, the pAlb (−3k) data were eliminated from Fig. 2E. This indicated that the pAlb (−1
k) was the best candidate to selectively express the two
proto-oncogenes in hepatocytes. Thus, the porcine promoter region
(−1,036 to −7 nt) was selected to induce hepatocyte-specific
expression.
Construction of overexpression vectors to
induce HCC in a pig model
For the final transgenic vectors, the Alb promoter
was transfected into the vectors that overexpress the two
proto-oncogenes (Fig. 3A). Two
overexpression vectors are represented in Fig. 3B and these were confirmed by the
digestion patterns in Fig. 3C. The
functional properties of the vectors were further confirmed with
RFP (Fig. 3D) and Myc (Fig. 3E) expression in the kidney and
liver cell lines. As expected, the two overexpressing vectors
controlled by the CMV presented strong RFP signals and high protein
levels of Myc in the cell lines compared with those of the
corresponding controls. The Alb promoter controlling the
proto-oncogene-expressing vectors induced RFP signals and displayed
Myc expression in the Hep G2 cells but not in the 293T cells. This
indicated that the transgenic vectors had functionally induced two
proto-oncogenes specifically in hepatocytes.
Generation of transgenic cell lines to be
used as the source of nuclear transfer
To generate transgenic cell lines for SCNT, the
transgenic vectors were linearized with a restriction enzyme
(NheI) and introduced into porcine fetal fibroblasts via a
liposome-mediated DNA delivery system. This delivery system avoided
unwanted side-effects, which may originate from the viral mediating
system. To aid the screening of a positive clone, the cytotoxicity
of neomycin was assayed using the fibroblasts; concentrations of
>30 mg/ml G-418 effectively eliminated (<20% survival rate)
non-transgenic cells within 3 days (Fig. 4A). The antibiotic-resistant clones
were screened for >3 weeks until colony formation occurred and
they were then further confirmed using PCR-based genotyping
(Fig. 4B). The integration rates
of the transgenic vectors into genomic DNA were >500 copies when
compared with the copy of the endogenous porcine albumin gene (data
not shown). Thus, three transgenic cell lines (TGF-α, Myc, and a
combination of the two) were generated and these can be used in
future SCNT to produce liver cancer porcine models.
Discussion
Liver cancer is a lethal disease and the
well-defined risk factors for HCC in humans include cirrhosis,
chronic hepatitis B and C viral infection, chronic alcohol
consumption and afatoxin-B1 intake. Animal models are widely used
to improve our understanding of HCC, in particular mouse models
(4). These mouse models include
the carcinogen-induced model, the implantation model, the
genetically engineered mouse, and the viral hepatocarcinogenesis
model, which are distinguished by etiological aspects (4). However, none of the currently
available mouse models meet all the criteria for the ideal animal
model, including biological, genetic, etiological and therapeutic
criteria (10).
The most frequently used method for creating a model
for HCC is carcinogen treatment, and numerous chemicals have been
shown to induce tumors in the mouse liver (11,12).
Hepato-carcinogens induce cancer via genotoxic and/or epigenetic
(or non-genotoxic) effects. The genotoxic carcinogens induce
genetic changes in the target cell, so that it develops into a
pre-neoplastic state. The epigenetic carcinogens stimulate the
pre-neoplastic state to evolve into a malignant neoplasm by
controlling cell proliferation, apoptosis and cell differentiation
without DNA modification (12–14).
Common hepatocarcinogens include diethylnitrosamine and
phenobarbital (4,15). These chemicals are either
administered to newborn mice in order to determine genotoxicity, or
for longer periods to induce epigenetic carcinogenesis (11,16).
Although carcinogen-induced mouse models for HCC are useful for
establishing an association between carcinogen exposure and
specific genetic changes, the influences of gender, age and the
genetic background of the mice on the predictability of HCC
development remain disadvantages of these models (17).
More than 80% of HCCs in humans are attributable to
infection with either the hepatitis B virus (HBV), the hepatitis C
virus (HCV) or infection with both (18). This virus-mediated HCC is
characteristically preceded by liver cirrhosis and may take more
than two decades to develop, implying that hepatocarcinogenesis
caused by viral hepatitis requires multiple steps of genetic
alterations (19). In addition,
HBV and HCV require the presence of human hepatocytes to induce
hepatitis, due to the stringent human tropism of these viruses
(20,21). Although viral hepatitis is the main
cause of HCC, the relatively long pathogenesis and the lack of the
virus inducing HCC in porcine models restrict us from using this
method of viral hepatocarcinogenesis.
Although the exact genetic events in
hepatocarcinogenesis are not clear, there is evidence that the p53,
Rb and Wnt/β-catenin pathways are involved (22,23).
Several transgenic mouse lines that are currently used to induce
the formation of HCCs are transgenic in one of these pathways
(4). Of the transgenic mice
expressing the Simian virus 40 large T-antigen that are directed to
the liver by the promoter of antithrombin-III, albumin and
α-1-antitrypsin, the majority developed hepatocarcinoma within one
year (5,24,25).
The T-antigen causes malignant transformation of the host cell
primarily by inactivating the tumor-suppressor genes; P53 and Rb
(26,27). A double transgenic mouse model
overexpressing TGF-α and Myc developed HCC substantially quicker
and at a higher rate than the single transgenic 8-month-old mice
(100% of males and 30% of females) (4). This gender-dependent carcinogenesis
is similar to the human etiology (4). Based on the mouse models, it is
expected that the transgenic cells originating from the pigs in the
current study will induce HCC at an early age.
Although surgical approaches such as liver resection
and transplantation are considered the most effective treatments to
cure HCC, a large portion of patients are unsuitable candidates for
these approaches due to the development of multicentric tumors,
extrahepatic metastases and early vascular invasion, in addition to
a shortage of donor organs, a high complication rate and
comorbidities (28–32). Local methods of tumor ablation
including TACE, percutaneous ethanol injection, radiofrequency
ablation (33–35), microwave coagulation therapy and
laser-induced thermotherapy are commonly used (36). TACE has become one of the most
common forms of interventional therapy due to its low systemic
toxicity and high therapeutic results (37–39).
Although several methods and devices for treating HCC have been
developed and applied in clinical settings, these are still limited
by the lack of an appropriate animal model. To replace the current
rodent model, we suggest a pig model, which has similar body size
and physiological aspects to humans.
Pigs are one of the major animal species used in
translational research and are being used as an alternative to the
dog and monkey as the non-rodent of choice in preclinical research
(7). Multiple technical procedures
for the use of pigs in translational and preclinical studies are
available, and numerous studies regarding the anatomy, physiology
and pathology of the pig are also available (40,41).
Pigs have been the preferred option as a model for surgical
training and research into methods including interventional
catheter techniques, complex trauma procedures and endoscopic
procedures. Pigs are also ideal animals for the development of
devices and techniques, and the US Food and Drug Administration has
previously accepted data from pigs (7). This supports the rationale for the
use of pigs as a liver cancer disease model and an alternative
choice of non-rodent species.
In the present study, transgenic cell lines that
contained two well-known proto-oncogenes (TGF-α and Myc) controlled
by porcine albumin promoter were generated, and they may induce HCC
in a porcine model. The expression of a combination of two
proto-oncogenes was adopted to maximize carcinogenesis, based on
previous mouse models (4). The
albumin promoter was the best candidate to selectively express the
genes in hepatocytes and it was demonstrated that the selected
porcine albumin promoter region was highly active in the liver cell
line (Hep G2) but not in the kidney cell line (293T). Although HCC
occurrence in the pig model was not demonstrated in the present
study, the current transgenic cells have the potential to generate
an HCC pig model to replace the classical rodent model, which has
multiple limitations.
Acknowledgements
The current study was supported by a grant (no.
PJ009069) from the Next-Generation BioGreen 21 Program, Rural
Development Administration, Korea.
References
|
1
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Li Y, Tang ZY and Hou JX: Hepatocellular
carcinoma: insight from animal models. Nat Rev Gastroenterol
Hepatol. 9:32–43. 2011. View Article : Google Scholar
|
|
3
|
Lencioni R: Chemoembolization for
hepatocellular carcinoma. Semin Oncol. 39:503–509. 2012. View Article : Google Scholar
|
|
4
|
Leenders MW, Nijkamp MW and Borel Rinkes
IH: Mouse models in liver cancer research: a review of current
literature. World J Gastroenterol. 14:6915–6923. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dubois N, Bennoun M, Allemand I, et al:
Time-course development of differentiated hepatocarcinoma and lung
metastasis in transgenic mice. J Hepatol. 13:227–239. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Santoni-Rugiu E, Nagy P, Jensen MR, et al:
Evolution of neoplastic development in the liver of transgenic mice
co-expressing c-myc and transforming growth factor-alpha. Am J
Pathol. 149:407–428. 1996.PubMed/NCBI
|
|
7
|
Swindle MM, Makin A, Herron AJ, et al:
Swine as models in biomedical research and toxicology testing. Vet
Pathol. 49:344–356. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jung EM, Kim YK, Lee GS, et al:
Establishment of inducible cAMP early repressor transgenic
fibroblasts in a porcine model of human type 1 diabetes mellitus.
Mol Med Rep. 6:239–245. 2012.PubMed/NCBI
|
|
9
|
Kim YK, Lee GS, Jung EM, et al: Generation
of fibroblasts overexpressing liver-specific PEPCK in a miniature
pig model of human type 2 diabetes mellitus. Mol Med Rep. 6:45–50.
2012.PubMed/NCBI
|
|
10
|
Hann B and Balmain A: Building ‘validated’
mouse models of human cancer. Curr Opin Cell Biol. 13:778–784.
2001.
|
|
11
|
Huff J, Cirvello J, Haseman J and Bucher
J: Chemicals associated with site-specific neoplasia in 1394
long-term carcinogenesis experiments in laboratory rodents. Environ
Health Perspect. 93:247–270. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wogan GN: Impacts of chemicals on liver
cancer risk. Semin Cancer Biol. 10:201–210. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gonzalez FJ: The peroxisome
proliferator-activated receptor alpha (PPARalpha): role in
hepatocarcinogenesis. Mol Cell Endocrinol. 193:71–79. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Williams GM: Chemicals with carcinogenic
activity in the rodent liver; mechanistic evaluation of human risk.
Cancer Lett. 117:175–188. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee GH: Paradoxical effects of
phenobarbital on mouse hepatocarcinogenesis. Toxicol Pathol.
28:215–225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen CJ, Yu MW and Liaw YF:
Epidemiological characteristics and risk factors of hepatocellular
carcinoma. J Gastroenterol Hepatol. 12:S294–308. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hirst GL and Balmain A: Forty years of
cancer modelling in the mouse. Eur J Cancer. 40:1974–1980.
2004.PubMed/NCBI
|
|
18
|
Parkin DM: The global health burden of
infection-associated cancers in the year 2002. Int J Cancer.
118:3030–3044. 2006.PubMed/NCBI
|
|
19
|
Fattovich G, Stroffolini T, Zagni I and
Donato F: Hepatocellular carcinoma in cirrhosis: incidence and risk
factors. Gastroenterology. 127(Suppl 1): S35–S50. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dandri M, Volz TK, Lütgehetmann M and
Petersen J: Animal models for the study of HBV replication and its
variants. J Clin Virol. 34(Suppl 1): S54–S62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kremsdorf D and Brezillon N: New animal
models for hepatitis C viral infection and pathogenesis studies.
World J Gastroenterol. 13:2427–2435. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Buendia MA: Genetics of hepatocellular
carcinoma. Semin Cancer Biol. 10:185–200. 2000. View Article : Google Scholar
|
|
23
|
Colnot S, Decaens T, Niwa-Kawakita M, et
al: Liver-targeted disruption of Apc in mice activates beta-catenin
signaling and leads to hepatocellular carcinomas. Proc Natl Acad
Sci USA. 101:17216–17221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kitagawa T, Hino O, Lee GH, et al:
Multistep hepatocarcinogenesis in transgenic mice harboring SV40
T-antigen gene. Princess Takamatsu Symp. 22:349–360.
1991.PubMed/NCBI
|
|
25
|
Sepulveda AR, Finegold MJ, Smith B, et al:
Development of a transgenic mouse system for the analysis of stages
in liver carcinogenesis using tissue-specific expression of SV40
large T-antigen controlled by regulatory elements of the human
alpha-1-antitrypsin gene. Cancer Res. 49:6108–6117. 1989.
|
|
26
|
Ahuja D, Sáenz-Robles MT and Pipas JM:
SV40 large T antigen targets multiple cellular pathways to elicit
cellular transformation. Oncogene. 24:7729–7745. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ali SH and DeCaprio JA: Cellular
transformation by SV40 large T antigen: interaction with host
proteins. Semin Cancer Biol. 11:15–23. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alsowmely AM and Hodgson HJ: Non-surgical
treatment of hepatocellular carcinoma. Aliment Pharmacol Ther.
16:1–15. 2002. View Article : Google Scholar
|
|
29
|
Durand F and Belghiti J: Liver
transplantation for hepatocellular carcinoma.
Hepatogastroenterology. 49:47–52. 2002.PubMed/NCBI
|
|
30
|
Maataoui A, Qian J, Vossoughi D, et al:
Transarterial chemoembolization alone and in combination with other
therapies: a comparative study in an animal HCC model. Eur Radiol.
15:127–133. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Poon RT, Fan ST, Tsang FH and Wong J:
Locoregional therapies for hepatocellular carcinoma: a critical
review from the surgeon’s perspective. Ann Surg. 235:466–486.
2002.PubMed/NCBI
|
|
32
|
Tang ZY: Treatment of hepatocellular
carcinoma. Digestion. 59:556–562. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Buscarini E and Buscarini L:
Radiofrequency thermal ablation with expandable needle of focal
liver malignancies: complication report. Eur Radiol. 14:31–37.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Denys AL, De Baere T, Kuoch V, et al:
Radio-frequency tissue ablation of the liver: in vivo and ex vivo
experiments with four different systems. Eur Radiol. 13:2346–2352.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee JM, Lee YH, Kim YK, et al: Combined
treatment of radiofrequency ablation and acetic acid injection: an
in vivo feasibility study in rabbit liver. Eur Radiol.
14:1303–1310. 2004.PubMed/NCBI
|
|
36
|
Sturm JW, Keese MA, Bönninghoff RG, et al:
Locally ablative therapies of hepatocellular carcinoma. Onkologie.
24(Suppl 5): 35–45. 2001.(In German).
|
|
37
|
Achenbach T, Seifert JK, Pitton MB, et al:
Chemoembolization for primary liver cancer. Eur J Surg Oncol.
28:37–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Llovet JM, Real MI, Montaña X, et al;
Barcelona Liver Cancer Group. Arterial embolisation or
chemoembolisation versus symptomatic treatment in patients with
unresectable hepatocellular carcinoma: a randomised controlled
trial. Lancet. 359:1734–1739. 2002. View Article : Google Scholar
|
|
39
|
Vogl TJ, Trapp M, Schroeder H, et al:
Transarterial chemoembolization for hepatocellular carcinoma:
volumetric and morphologic CT criteria for assessment of prognosis
and therapeutic success - results from a liver transplantation
center. Radiology. 214:349–357. 2000.
|
|
40
|
Writing Group Members. Lloyd-Jones D,
Adams RJ, Brown TM, et al; American Heart Association Statistics
Committee and Stroke Statistics Subcommittee. Heart disease and
stroke statistics - 2010 update: a report from the American Heart
Association. Circulation. 121:e46–e215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Swindle MM, Smith AC and Hepburn BJ: Swine
as models in experimental surgery. J Invest Surg. 1:65–79. 1988.
View Article : Google Scholar : PubMed/NCBI
|