Introduction
Acute kidney injury (AKI) induced by renal ischemia
leads to significant morbidity and mortality (1). Studies have suggested that apoptotic,
autophagic and necrotic cell death are involved in AKI caused by
renal ischemia (2–5). Despite major advances in the
understanding of the molecular mechanisms associated with ischemic
AKI, treatment largely remains supportive and outcomes have not
improved during the past 50 years (6). Therefore, examining the molecular
mechanisms of AKI induced by renal ischemia has become increasingly
important.
Cell death was initially divided into apoptosis,
autophagy and necrosis according to morphological appearance
(7). Apoptosis refers to an active
programmed cell death, is executed by intrinsic or extrinsic
pathways in response to various cell death stimuli and is regulated
by a specific class of compounds, including the caspase cascade
(8). Apoptosis is characterized by
cell shrinkage, nuclear and cytoplasmic condensation, DNA
fragmentation and the formation of apoptosomes (9). Autophagy is an important mechanism to
maintain cell homeostasis and provides a pro-survival function
under starvation conditions. Distinct from apoptosis, necrosis is
considered a passive cell death program and is commonly viewed as
an accidental and unregulated event. Necrosis is morphologically
characterized by cellular swelling, swelling of organelles, plasma
membrane rupture and loss of intracellular contents (10).
A relatively new form of necrosis has been
characterized as ‘necroptosis’ (11,12).
Necroptosis is a newly identified type of programmed cell death
initiated by the activation of apoptosis induced by binding of
tumor necrosis factor (TNF) or Fas to its receptor, referred to as
a regulated form of necrosis, which is dependent on the
serine-threonine kinase receptor-interacting protein 1 (RIP1)
(13,14). Necroptosis exhibits morphological
characteristics, which resemble necrosis and can be specifically
inhibited by a small molecule, necrostatin-1 (Nec-1), which targets
RIP1 (15). As a newly identified
form of cell death, intense studies performed in recent years have
demonstrated that necroptosis is important in immune system
regulation, tissue injury and cellular responses to multiple
stresses (16–19).
Necroptosis is a potential therapeutic target for
treating numerous diseases. The present study examined whether
necroptosis was present in the human proximal tubular epithelial
cell line HK-2 following ischemic injury induced by adenosine
triphosphate (ATP) depletion and evaluated the effect of Nec-1, an
inhibitor of RIP1, which specifically inhibits necroptosis, in HK-2
cell injury in an in vitro ATP-depleted renal ischemia
model. In addition, the present study assessed whether Nec-1
inhibited autophagy, which is a caspase-independent process
activated during necroptosis.
Materials and methods
Cell culture
HK-2 cells were obtained from the American Tissue
Culture Collection (Rockville, MD, USA) and maintained in a mixture
of Ham’s F12 and Dulbecco’s modified Eagle’s medium (DMEM:F12;
Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum, 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco-BRL)
in a humidified atmosphere of 5% CO2 and 95% air at
37°C. The cells were seeded at an appropriate cell density for
different assays and left to grow to 80% confluence, and then cell
synchronization was routinely performed by incubating cells in
serum-free medium for 24 h prior to every experiment. Finally, the
cells were exposed to different experimental conditions.
ATP depletion
ATP depletion, an established model of renal
ischemia (20,21), was induced by exposing cells to
prewarmed glucose-free DMEM that contained the electron transport
chain inhibitor antimycin A (10 μM) at 37°C in the presence of 5%
CO2 as previously described (22). Glucose removal was required in
order to prevent cellular anaerobic glycolysis. The cells were
subjected to 30 min, 1 and 2 h of energy deprivation,
respectively.
Cell treatment
Cells were divided into 12 groups as follows: i)
Control group: Cells were maintained in DMEM:F12 supplemented with
10% FBS for 2 h; ii) TNF-α group: Cells were treated with 10 ng/ml
TNF-α (Sigma-Aldrich, St. Louis, MO, USA) for 2 h; iii) TNF-α +
z-VAD-fmk group: Cells were treated with 10 ng/ml TNF-α and 50 μM
of z-VAD-fmk (Sigma-Aldrich) for 2 h. iv) TNF-α + z-VAD-fmk +
antimycin A (Sigma-Aldrich) group: Cells were treated with 10 ng/ml
TNF-α and 50 μM z-VAD-fmk for 2 h, and with 10 μM antimycin A for
0.5, 1 and 2 h, respectively. v) Nec-1 + control group: Cells were
pretreated with 50 μM Nec-1 (Cell Signaling Technology, Shanghai,
China) for 6 h and then continuously maintained in DMEM:F12
supplemented with 10% FBS for 2 h; vi) Nec-1 + TNF-α group: Cells
were pretreated with 50 μM Nec-1 for 6 h and then continuously
treated with 10 ng/ml TNF-α for 2 h; vii) Nec-1 + TNF-α + z-VAD-fmk
group: Cells were pretreated with 50 μM Nec-1 for 6 h and then
continuously treated with 10 ng/ml TNF-α and 50 μM z-VAD-fmk for 2
h; viii) Nec-1 + TNF-α + z-VAD-fmk + antimycin A group: Cells were
pretreated with 50 μM of Nec-1 for 6 h and then continuously
treated with 10 ng/ml TNF-α and 50 μM z-VAD-fmk for 2 h, and with
10 μM antimycin A for 0.5, 1 and 2 h, respectively.
Cellular morphological assessment
A phase-contrast inverted microscope and
transmission electron microscope were used to survey the changes in
cellular morphology. The cells were seeded in 25 mm culture flasks
(Coning Inc., Corning, NY, USA) at a cell density of 105
cells. At the end of the incubation period, cells were visualized
and images were captured by a phase-contrast inverted microscope
(IX71; Olympus, Tokyo, Japan) with a digital camera (Olympus DP72,
U-TV0.63XC; Olympus). A transmission electron microscope
(JEM-100CX; JEOL Ltd., Tokyo, Japan) was used to observe and
capture images of the ultrastructure of cells. The cells were
trypsinized and washed with phosphate-buffered saline, and then
cell pellets were fixed with 2% glutaraldehyde in 0.15 mM
cacodylate buffer (pH 7.2) at 4°C for 1 h, and washed in 0.15 mM
cacodylate buffer (3X; 5 min). Following that, the cell pellets
were fixed in 1% osmium tetroxide at 4°C for 30 min, rinsed with
dH2O at 4°C (3X; 2 min) and stained with 1% aqueous
uranyl acetate at 4°C for 30 min. Finally, cell pellets were
dehydrated stepwise with ethanol and embedded in resin (Epon 812)
for 12 h. Following 12 h in the oven at 60°C for polymerization,
the samples were cut into extremely thin sections (1 μm), collected
and placed on a small circular metal grid prior to being
viewed.
Cell viability assay (CCK-8 assay)
HK-2 cells under different experimental conditions
were harvested using the Cell Counting kit-8 (CCK-8; Dojindo
Laboratories, Kumamoto, Japan) assay to determine the number of
viable cells according to the manufacturer’s instructions. Briefly,
cells were seeded on a 96-well plate at a concentration of
104 cells per well and then cells were treated with
various experimental conditions as mentioned above. Following
incubation, 10 μl of CCK-8 solution was added to each well and the
96-well plate was continuously incubated at 37°C for 4 h. Then the
OD value for each well was read at a wavelength of 450 nm to
determine the cell viability using a microplate reader (Model 550;
Bio-Rad, Hercules, CA, USA). The assay was repeated three times.
The cell viability was calculated using the following formula: Cell
viability (%) = OD (experiment) − OD (blank) / OD (control) − OD
(blank) × 100%.
Western blot analysis
HK-2 cells under different experimental conditions
were lysed in lysis buffer. The total protein concentration from
the resultant supernatant was determined by the bicinchoninic acid
protein assay (Beyotime Institute of Biotechnology, Nantong,
Jiangsu, China). An aliquot of cell lysates containing 30 μg of
protein were separated by SDS-PAGE and transferred onto a
polyvinylidene difluoride membrane (Immobilon-P; Millipore,
Billerica, MA, USA) by electroblotting. The membranes were
incubated overnight at 4°C with rabbit anti-LC3B antibody (L7453;
Sigma, St Louis, MO, USA). Following washing, the secondary
antibody (Thermo Fisher Scientific, Waltham, MA, USA) was added and
incubated for 1 h at room temperature. The protein bands were
visualized by Enhanced Chemiluminescence Plus (Amersham, Arlington
Heights, IL, USA) and then exposed to X-ray film (Kodak, Rochester,
NY, USA). The bands of the resulting autoradiographs were
quantified densitometrically using Bandscan software (version 5.0;
Funglyn Biotech, Toronto, ON, Canada). Protein expression was
quantified as the ratio of specific band to GAPDH.
Statistical analysis
All values are expressed as the mean ± standard
error. Statistical analysis was performed using the statistical
package SPSS for Windows version 13.0 (SPSS, Inc., Chicago, IL,
USA). Multiple comparisons among the groups were conducted by
one-way analysis of variance with Tukey’s tests for post-hoc
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
Nec-1 and z-VAD-fmk ameliorate
morphological abnormality of HK-2 cells following TNF-α stimulation
and ATP depletion
ATP depletion is usually applied in in vitro
experiments to mimic the renal injury process of
ischemia-reperfusion injury (IRI) (20,21).
HK-2 cells were treated with 10 ng/ml TNF-α and 50 μmol/l z-VAD-fmk
for 2 h. Following that, the ATP depleter antimycin A (10 mM) was
added into the glucose-free medium to incubate the HK-2 cells for
different periods of time. Fig.
1A–B shows morphological changes determined by a phase-contrast
inverted microscope and a transmission electron microscope,
respectively. Fig. 1Aa–d represent
the control group, TNF-α + z-VAD-fmk + antimycin A (1 h) group, the
Nec-1 + control group and the Nec-1 + TNF-α + z-VAD-fmk + antimycin
A (1 h) group, respectively. The morphological abnormalities,
including cellular swelling, swelling of organelles and plasma
membrane rupture, were significantly ameliorated in the Nec-1 +
TNF-α + z-VAD-fmk + antimycin A (1 h) group compared with the TNF-α
+ z-VAD-fmk + antimycin A (1 h) group. Fig. 1Aa–c represents the control group,
TNF-α + z-VAD-fmk + antimycin A (1 h) group and the Nec-1 + TNF-α +
z-VAD-fmk + antimycin A (1 h) group, respectively. Similarly, these
results are also observed using a transmission electron microscope
in the Nec-1 + TNF-α + z-VAD-fmk + antimycin A (1 h) group compared
with the TNF-α + z-VAD-fmk + antimycin A (1 h) group.
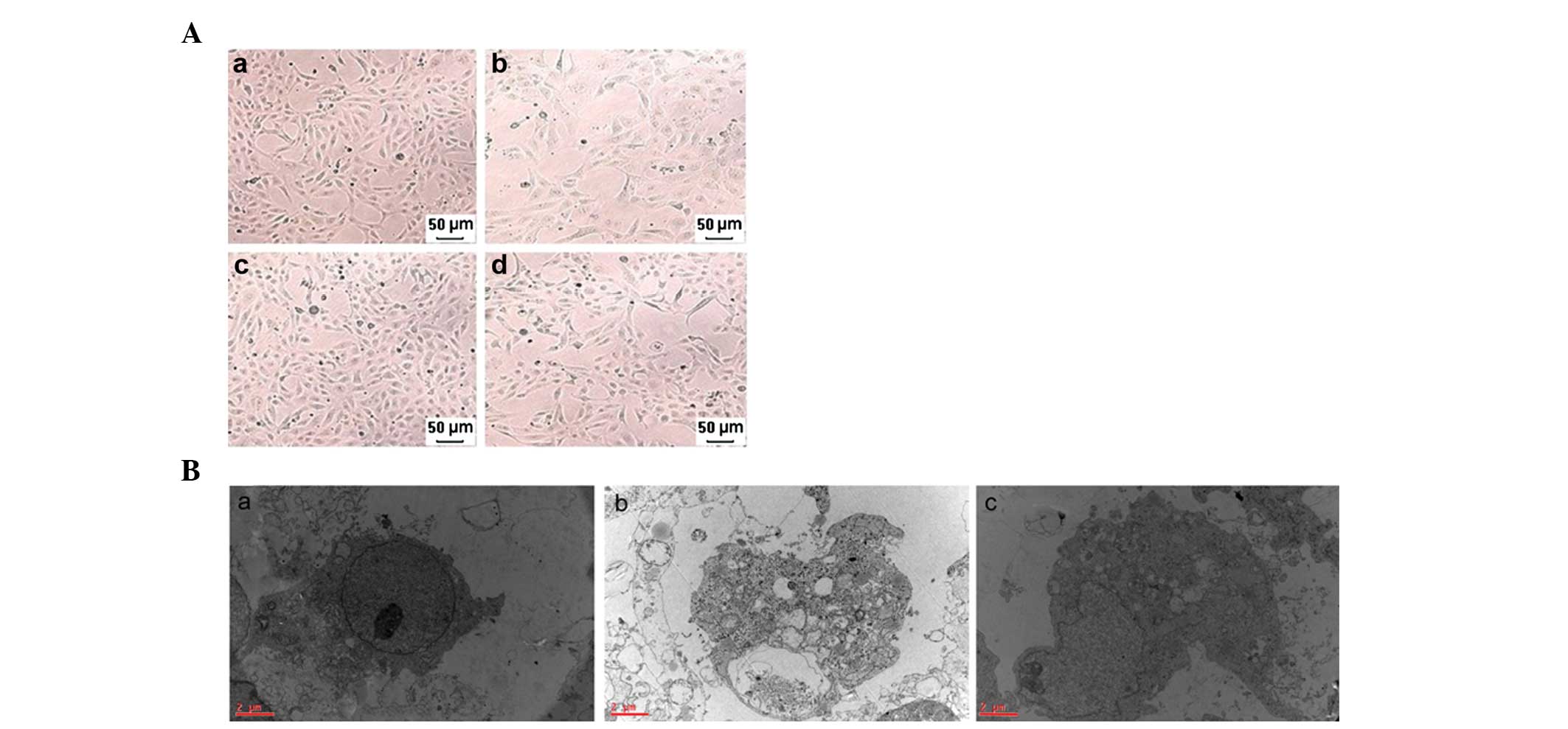 | Figure 1Morphology of HK-2 cells determined by
a phase-contrast inverted microscope and a transmission electron
microscope. (A) Representative phase-contrast inverted microscope
images of HK-2 cells: (a) Control group, (b) TNF-α + z-VAD-fmk +
antimycin A (1 h) group, (c) Nec-1 + control group, (d) Nec-1 +
TNF-α + z-VAD-fmk + antimycin A (1 h) group (magnification, ×100).
(B) Representative electron micrographs of HK-2 cells
(magnification, ×20,000): (a) Control group, (b) TNF-α + z-VAD-fmk
+ antimycin A (1 h) group, (c) Nec-1 + TNF-α + z-VAD-fmk +
antimycin A (1 h) group. Note the prominent nuclear swelling, loss
of nuclear condensation and loss of mitochondrial and endoplasmic
reticulum mass as hallmarks of programmed necrosis. Bar=2 μm.
Nec-1, necrostatin-1; TNF-α, tumor necrosis factor-α. |
Nec-1 increased cell viability in HK-2
cells following TNF-α stimulation and ATP depletion
As shown in Fig. 2,
cell viability was significantly decreased in the TNF-α group
(50.3±1.4%), TNF-α + z-VAD-fmk group (43.7±1.5%), TNF-α + z-VAD-fmk
+ antimycin A (30 min) group (49.7±1.4%), TNF-α + z-VAD-fmk +
antimycin A (1 h) group (30.8±0.8%) and the TNF-α + z-VAD-fmk +
antimycin A (2 h) group (15.1±0.6%) compared with the control group
(100±0.0%), respectively. Pretreatment with Nec-1 (50 μM) had no
effect on cell viability in the control group (95.6±3.0%), TNF-α
group (49.5±1.2%) and TNF-α + z-VAD-fmk group (43.0±1.4%),
respectively. This was apparent by comparison of the Nec-1 +
control group (95.6±3.0%) with the control group (100±0.0%;
P>0.05); the Nec-1 + TNF-α group (49.5±1.2%) with the TNF-α
group (50.3±1.4%; P>0.05) and the Nec-1 + TNF-α z-VAD-fmk group
(43.0±1.4%) with the TNF-α + z-VAD-fmk group (43.7±1.5%;
P>0.05). Following treatment with 10 μM antimycin A for 0.5, 1
and 2 h, respectively, Nec-1 (50 μM) pretreatment significantly
increased cell viability from 30.8±0.8% (TNF-α + z-VAD-fmk +
antimycin A; 1 h group) to 69.4±0.8% (Nec-1 + TNF-α + z-VAD-fmk +
antimycin A; 1 h group; P<0.05). However, no difference in cell
viability in the Nec-1 + TNF-α + z-VAD-fmk + antimycin A (30 min)
group (47.3±0.9%) and the Nec-1 + TNF-α + z-VAD-fmk + antimycin A
(2 h) group (12.4±0.5%) compared with the TNF-α + z-VAD-fmk +
antimycin A (30 min) group (49.7±1.4%) and the TNF-α + z-VAD-fmk +
antimycin A (2 h) group (15.1±0.6%; P>0.05) was observed,
respectively, as shown in Fig.
2.
 | Figure 2Viability of HK-2 cells measured by
the CCK-8 assay and expressed as percentages of the control. Data
are expressed as the mean ± standard error of four independent
experiments. *P<0.05, versus control group;
#P<0.05, versus TNF-α + z-VAD-fmk + antimycin A (1 h)
group. Con, control group; T, TNF-α group; T + Z, TNF-α + z-VAD-fmk
group; T + Z + A1, TNF-α + z-VAD-fmk + antimycin A (30 min) group;
T + Z + A2, TNF-α + z-VAD-fmk + antimycin A (1 h) group; T + Z +
A3, TNF-α + z-VAD-fmk + antimycin A (2 h) group; Nec-1 + con, Nec-1
+ control group; Nec-1 + T, Nec-1 + TNF-α group; Nec-1 + T + Z,
Nec-1 + TNF-α + z-VAD-fmk group; Nec-1 + T + Z +A1, Nec-1 + TNF-α +
z-VAD-fmk + antimycin A (30 min) group; Nec-1 + T + Z + A2, Nec-1 +
TNF-α + z-VAD-fmk + antimycin A (1 h) group; Nec-1 + T + Z+A3,
Nec-1 + TNF-α + z-VAD-fmk + antimycin A (2 h) group; CCK-8, Cell
Counting kit-8; Nec-1, necrostatin-1; TNF-α, tumor necrosis
factor-α. |
Nec-1 inhibits autophagy following TNF-α
stimulation and ATP depletion
In order to further examine the specificity of Nec-1
in renal ischemia HK-2 cells, the present study also assessed
whether autophagy, which is a caspase-independent process, is
inhibited, and is directly associated with the conversion of the
microtubule associated protein light chain (LC)-3 (I) to LC-3 (II).
In HK-2 cells subjected to TNF-α and z-VAD-fmk treatment in the
presence of ATP depletion for 1 h, it was demonstrated that the
changes of the protein levels of the LC3-II/I ratio were
significantly increased (1.49±0.11 vs control 0.70±0.03;
P<0.05); however, when the cells were pretreated with Nec-1,
this increase was markedly inhibited (0.68±0.02 vs 1.49±0.11;
P<0.05). Similarly, besides the increase in the LC3-II/I ratio,
the LC3-II/GAPDH ratio under Nec-1 treatment was also significantly
inhibited in the Nec-1 + TNF-α + z-VAD-fmk + antimycin A (1 h)
group (0.73±0.03) compared with the TNF-α + z-VAD-fmk + antimycin A
(1 h) group (1.72±0.10). No difference in the LC3-II/I ratio and
LC3-II/GAPDH ratio in the Nec-1 + TNF-α group (0.68±0.03,
0.67±0.01), Nec-1 + TNF-α + z-VAD-fmk group (1.08±0.04, 1.36±0.09),
Nec-1 + TNF-α + z-VAD-fmk + antimycin A (30 min) group (1.18±0.04,
1.33±0.09) and the Nec-1 + TNF-α + z-VAD-fmk + antimycin A (2 h)
group (0.87±0.04, 0.9±0.07) was observed compared with the TNF-α
group (0.65±0.03, 0.68±0.01), TNF-α + z-VAD-fmk group (1.14±0.07,
1.52±0.09), TNF-α + z-VAD-fmk + antimycin A (30 min) group
(1.13±0.02, 1.36±0.04) and the TNF-α + z-VAD-fmk + antimycin A (2
h) group (1.04±0.07, 1.07±0.06; P>0.05) respectively, as shown
in Fig. 3.
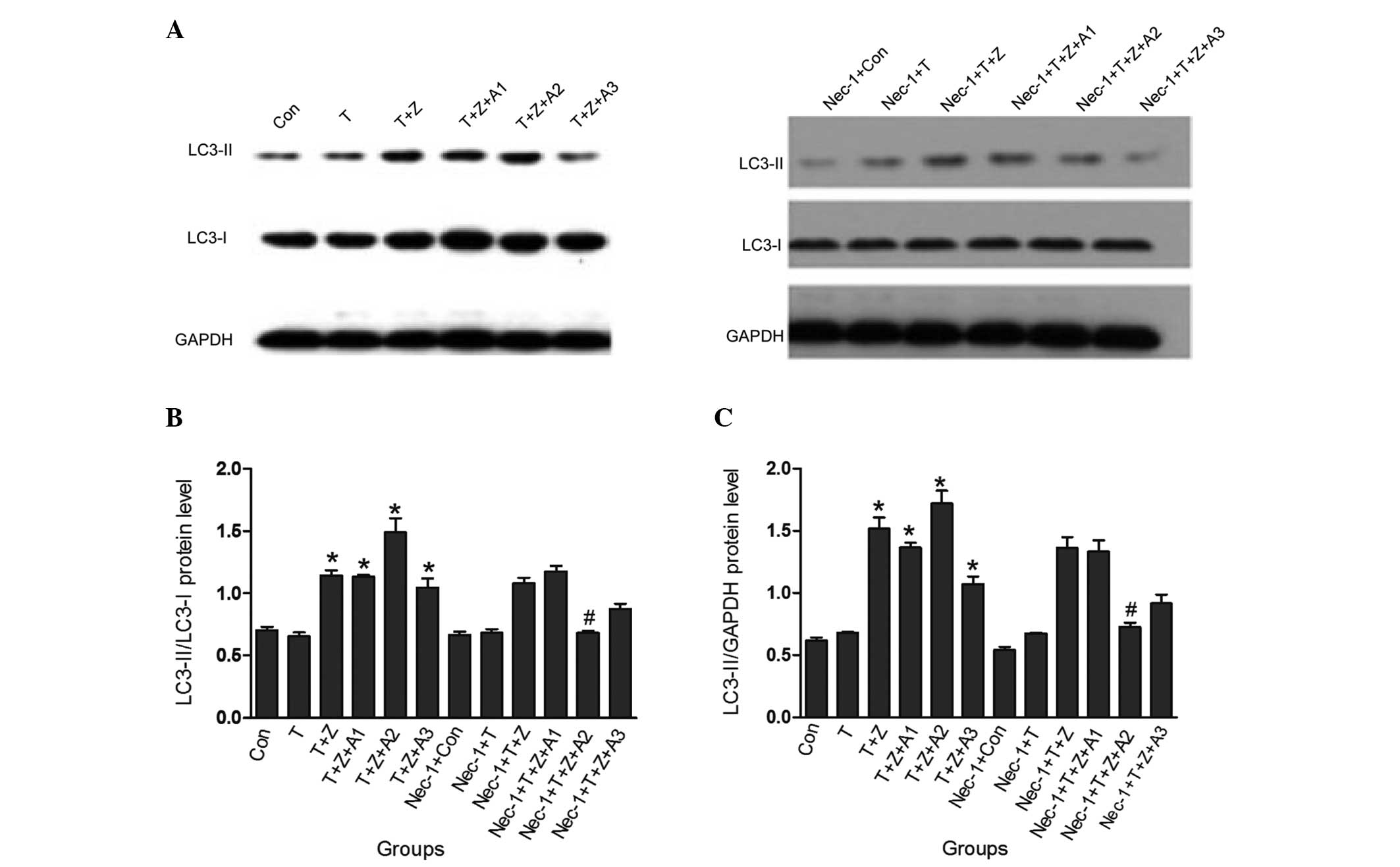 | Figure 3Nec-1 inhibits autophagy following
TNF-α stimulation and ischemic renal injury. TNF-α (10 ng/ml) and
z-VAD-fmk (50 μM) and/or antimycin A1 (10 μM) were added for the
indicated time periods, and the total cell lysates were prepared
and immunoblotted with LC3 and GAPDH antibodies. Blots are
representatives of three independent experiments. The protein
levels of LC3-I, LC3-II and GAPDH were quantified by densitometry,
and LC3-II/I and LC3-II/GAPDH ratios were calculated.
*P<0.05, versus control group; #P<0.05,
versus TNF-α + z-VAD-fmk + antimycin A (1 h) group. Con, control
group; T, TNF-α group; T + Z, TNF-α + z-VAD-fmk group; T + Z + A1,
TNF-α + z-VAD-fmk + antimycin A (30 min) group; T + Z + A2, TNF-α +
z-VAD-fmk + antimycin A (1 h) group; T + Z + A3, TNF-α + z-VAD-fmk
+ antimycin A (2 h) group; Nec-1 + con, Nec-1 + control group;
Nec-1 + T, Nec-1 + TNF-α group; Nec-1 + T + Z, Nec-1 + TNF-α +
z-VAD-fmk group; Nec-1 + T + Z + A1, Nec-1 + TNF-α + z-VAD-fmk +
antimycin A (30 min) group; Nec-1 + T + Z + A2, Nec-1 + TNF-α +
z-VAD-fmk + antimycin A (1 h) group; Nec-1 + T + Z + A3, Nec-1 +
TNF-α + z-VAD-fmk + antimycin A (2 h) group; Nec-1, necrostatin-1;
TNF-α, tumor necrosis factor-α; LC-3, protein light chain 3. |
Discussion
The present study investigated whether
caspase-independent cell death (necroptosis) was present in
cultured HK-2 cells subjected to energy depletion, and whether the
addition of Nec-1, a highly specific receptor-interacting protein
kinase 1 inhibitor, improves cell survival following TNF-α
stimulation and ATP depletion in an HK-2 ischemic injury model. The
present study supports the existence of a novel form of
caspase-independent programmed necrosis involving the activation of
autophagy in an in vitro model of renal ischemia. Nec-1, a
specific inhibitor of necroptosis, increased cell viability in
cultured HK-2 cells following TNF-α stimulation and ATP
depletion.
AKI induced by renal ischemia is a common clinical
complication characterized by an abrupt decrease in the glomerular
filtration rate. Despite supportive care, including renal
replacement therapy, the five-year mortality rate following AKI
remains at ~50% (19). Limited
understanding of the cellular mechanisms of AKI complicates the
development of an effective treatment. Therefore, examining the
molecular mechanisms of AKI has become increasingly important.
Cell death is essential in organ development, tissue
homeostasis and degenerative diseases. Three major types of cell
death have been described, including apoptosis (23), autophagy (24) and necrosis (25), based on their distinct cell
morphology. AKI involves these mechanisms of cell death (2–5).
Apoptotic cell death is characterized by the activation of caspases
and the formation of apoptotic bodies (8,9),
whereas autophagic cell death is differentiated by large-scale
sequestration of portions of the cytoplasm in autophagosomes,
giving the cell a characteristic vacuolated appearance (26). Distinct from apoptosis, necrosis is
morphologically characterized by an early onset of plasma membrane
permeabilization, which causes cells to swell and finally rupture,
spilling their intracellular contents into the extracellular milieu
(10). For numerous years,
necrosis was stereotyped as being a form of death occurring in an
unregulated manner, resulting from excessive stress. However, an
increasing body of evidence from previous years indicated that
necrosis may also be executed by regulated mechanisms (13). This regulated non-apoptotic cell
death was recently termed necroptosis (11), which is caspase-independent and
exhibits the morphological features of necrosis (early membrane and
organelle swelling followed by cell lysis). Necroptosis involves
Fas/TNF-α death domain receptor activation and inhibition of RIP1
kinase, and it has been suggested that it may contribute to the
development of neurological and myocardial diseases (11,18).
In the present study, cell viability was
significantly decreased in HK-2 cells following TNF-α stimulation
and ATP depletion. The cell viability decreased in a time-dependent
manner. When the cells were pretreated with Nec-1 for 6 h, the cell
viability was significantly improved [Nec-1 + TNF-α + z-VAD-fmk +
antimycin A (1 h) group, 69.4±1.7% vs. the TNF-α + z-VAD-fmk +
antimycin A (1 h) group, 30.8±1.7%; P<0.05] in HK-2 cells
following the addition of TNF-α and z-VAD, and ATP depletion. This
result indicated that necroptosis was present in HK-2 cell injury
induced by TNF-α stimulation and energy depletion (1 h). In the
present study, it was noted that Nec-1 pretreatment did not
increase cell viability when HK-2 cells were subjected to
ATP-depleted ischemic injury for 2 h. This indicated that
necroptosis had an early onset and thus drug interventions
targeting the mechanisms of necroptosis need to be used as early as
possible. In addition, Nec-1 pretreatment did not increase cell
viability when HK-2 cells were subjected to TNF-α and z-VAD-fmk
treatment. Pretreatment with Nec-1 only demonstrated a protective
effect on ATP-depleted HK-2 cells treated with TNF-α and z-VAD-fmk.
This may possibly be explained by improved stimulation of the
pathogenic background of developing AKI. Similarly, morphological
abnormalities, including cellular swelling, swelling of organelles
and plasma membrane rupture of HK-2 cells determined by microscopy
were markedly attenuated in HK-2 cell ischemic injury following the
use of Nec-1. This result further supported the evidence that the
presence of necroptosis and Nec-1 had a protective effect on the
HK-2 cell ischemic injury model.
In certain studies, necroptosis was characterized by
the activation of autophagy, which in turn contributes to cell
death (11,27–29).
Under conditions of renal ischemic injury, it remains to be
elucidated whether necroptosis concomitantly activated autophagy in
HK-2 cells. Therefore, the expression levels of LC-3 (II), a marker
of autophagy, and the expression change following the use of Nec-1
in HK-2 cell ischemic injury induced by TNF-α stimulation and
energy depletion were investigated. The present study demonstrated
that the LC3 II/I ratio was increased in the TNF-a + z-VAD-fmk +
antimycin A (1 h) group, while the increased LC3 II/I ratio was
markedly inhibited following treatment with Nec-1. Since the ratio
of LC3-II to GAPDH is an accurate indicator of autophagy (30), the ratio of LC3-II/GAPDH in HK-2
cells was also analyzed. The treatment with Nec-1 for 1 h
significantly decreased the ratio of LC3-II to GAPDH relative to
the control (Nec-1 + TNF-α + z-VAD-fmk + antimycin A group
0.73±0.03 vs. TNF-α + z-VAD-fmk + antimycin A group 1.72±0.10;
P<0.05). These data closely parallel the activity profile of
Nec-1 in necroptotic cells and suggest that Nec-1 targets a
caspase-independent necroptotic pathway involving autophagy in
vitro.
In conclusion, the present study confirmed that
necroptosis, a caspase-independent programmed cell death, is
involved in renal epithelial cell damage. For the first time, to
the best of our knowledge, the present study revealed that Nec-1
suppressed renal epithelial cell damage in a TNF-α-stimulated and
ATP-depleted renal ischemia model. In addition, it was noted that
Nec-1 targeted a caspase-independent necroptotic pathway
concomitantly involving autophagy. The results of the present study
suggested that necroptosis may be an important emerging mode of
cell death in acute renal ischemia. The therapeutic effects
demonstrated with Nec-1 support its use as a novel renal-protective
agent that targets necrotic cell death mechanisms. The inhibition
of necroptosis may be a new strategy for the development of
cytoprotective agents in AKI. However, further investigations are
required in order to clarify the mechanisms by which necroptosis
occurs in AKI.
Acknowledgements
This study was supported by the National Natural
Science Foundation (no. 81170683) and National Key Technology
R&D Program (No. 2011BAI10B08), National Clinical Key Specialty
Construction Preparatory Projects.
References
|
1
|
Nash K, Hafeez A and Hou S:
Hospital-acquired renal insufficiency. Am J Kidney Dis. 39:930–936.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Havasi A and Borkan S: Apoptosis and acute
kidney injury. Kidney Int. 80:29–40. 2011. View Article : Google Scholar
|
|
3
|
Jiang M, Wei Q, Dong G, Komatsu M, Su Y
and Dong Z: Autophagy in proximal tubules protects against acute
kidney injury. Kidney Int. 82:1271–1283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Padanilam BJ: Cell death induced by acute
renal injury: a perspective on the contributions of apoptosis and
necrosis. Am J Physiol Renal Physiol. 284:F608–F627.
2003.PubMed/NCBI
|
|
5
|
Matsuyama M, Hayama T, Funao K, Naganuma
T, Kawahito Y, Sano H, Chargui J, Touraine JL, Nakatani T and
Yoshimura R: The effect of neutrophil elastase inhibitor on acute
tubular necrosis after renal ischemia-reperfusion injury. Mol Med
Rep. 1:489–492. 2008.PubMed/NCBI
|
|
6
|
Ympa YP, Sakr Y, Reinhart K and Vincent
JL: Has mortality from acute renal failure decreased? A systematic
review of the literature. Am J Med. 118:827–832. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kroemer G, Galluzzi L, Vandenabeele P, et
al: Classification of cell death: recommendations of the
Nomenclature Committee on Cell Death 2009. Cell Death Differ.
16:3–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galluzzi L, Joza N, Tasdemir E, Maiuri MC,
Hengartner M, Abrams JM, Tavernarakis N, Penninger J, Madeo F and
Kroemer G: No death without life: vital function of apoptotic
effectors. Cell Death Differ. 15:1113–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kitanaka C and Kuchino Y:
Caspase-independent programmed cell death with necrotic morphology.
Cell Death Differ. 6:508–515. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu W, Liu P and Li J: Necroptosis: an
emerging form of programmed cell death. Crit Rev Oncol Hematol.
82:249–258. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galluzzi L and Kroemer G: Necroptosis: a
specialized pathway of programmed necrosis. Cell. 135:1161–1163.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin J, Jin X, Qian C, Ruan Y and Jiang H:
Signaling network of OSW-1-induced apoptosis and necroptosis in
hepatocellular carcinoma. Mol Med Rep. 7:1646–1650. 2013.PubMed/NCBI
|
|
15
|
Degterev A, Hitomi J, Germscheid M, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han J, Zhong CQ and Zhang DW: Programmed
necrosis: backup to and competitor with apoptosis in the immune
system. Nat Immunol. 12:1143–1149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Smith CC and Yellon DM: Necroptosis,
necrostatins and tissue injury. J Cell Mol Med. 15:1797–1806. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oerlemans MI, Liu J, Arslan F, den Ouden
K, van Middelaar BJ, Doevendans PA and Sluijter JP: Inhibition of
RIP1-dependent necrosis prevents adverse cardiac remodeling after
myocardial ischemia- reperfusion in vivo. Basic Res Cardiol.
107:2702012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Linkermann A, Bräsen JH, Himmerkus N, Liu
S, Huber TB, Kunzendorf U and Krautwald S: Rip1
(receptor-interacting protein kinase 1) mediates necroptosis and
contributes to renal ischemia/reperfusion injury. Kidney Int.
81:751–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lieberthal W, Menza SA and Levine JS:
Graded ATP depletion can cause necrosis or apoptosis of cultured
mouse proximal tubular cells. Am J Physiol. 274:F315–F327.
1998.PubMed/NCBI
|
|
21
|
Ruchalski K, Mao H, Singh SK, Wang Y,
Mosser DD, Li F, Schwartz JH and Borkan SC: HSP72 inhibits
apoptosis-inducing factor release in ATP-depleted renal epithelial
cells. Am J Physiol Cell Physiol. 285:C1483–C1493. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brooks C, Ketsawatsomkron P, Wang J, Wang
CY, Yu FS and Dong Z: Acidic pH inhibits ATP depletion-induced
tubular cell apoptosis by blocking caspase-9 activation in
apoptosome. Am J Physiol Renal Physiol. 289:F410–F419. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ueda N, Kaushal GP and Shah SV: Apoptotic
mechanisms in acute renal failure. Am J Med. 108:403–415. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsujimoto Y and Shimizu S: Another way to
die: autophagic programmed cell death. Cell Death Differ.
12:1528–1534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ha HC and Snyder SH: Poly(ADP-ribose)
polymerase is a mediator of necrotic cell death by ATP depletion.
Proc Natl Acad Sci USA. 96:13978–13982. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baehrecke EH: Autophagy: dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bell BD, Leverrier S, Weist BM, Newton RH,
Arechiga AF, Luhrs KA, Morrissette NS and Walsh CM: FADD and
caspase-8 control the outcome of autophagic signaling in
proliferating T cells. Proc Natl Acad Sci USA. 105:16677–16682.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bonapace L, Bornhauser BC, Schmitz M,
Cario G, Ziegler U, Niggli FK, Schäfer BW, Schrappe M, Stanulla M
and Bourquin JP: Induction of autophagy-dependent necroptosis is
required for childhood acute lymphoblastic leukemia cells to
overcome glucocorticoid resistance. J Clin Invest. 120:1310–1323.
2010. View
Article : Google Scholar
|
|
29
|
Rosenbaum DM, Degterev A, David J,
Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J and Savitz SI:
Necroptosis, a novel form of caspase-independent cell death,
contributes to neuronal damage in a retinal ischemia-reperfusion
injury model. J Neurosci Res. 88:1569–1576. 2010.PubMed/NCBI
|
|
30
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2008. View Article : Google Scholar : PubMed/NCBI
|














