Introduction
Cardiac conotruncal defects (CTDs) are cardiac
outflow tract defects that occur during the embryonic development
of complex congenital heart malformations. Examples of CTDs
include: Tetralogy of Fallot (TOF); pulmonary atresia with
ventricular septal defect (PA/VSD); double outlet of right
ventricle (DORV); transposition of the great arteries (TGA);
persistent truncus arteriosus (PTA); and interrupted aortic arch
(IAA). CTDs accounted for 25–33% of all congenital heart defects
and 70% of cyanotic congenital heart disease in 1996 (2). CTDs are the main cause of complex
cardiac malformations, mortality and overall serious harm to health
in infants, creating a heavy financial burden for families and
society (1,2).
Studies have shown that genetic factors are pivotal
in the pathogenesis of cardiac CTDs, but the mode of inheritance,
the penetrance, and the identities of susceptibility genes are not
yet clear (3,4). Previous studies have shown that the
22q11.2 microdeletion is associated with 75–85% of cardiac CTDs,
but only 6.1–17.9% of patients with nonsyndromic CTD have 22q11.2
microdeletions (5,6). Therefore, the genetic mechanisms of
>80% of nonsyndromic cardiac conotruncal malformations are
unknown. As a result of this, the screening of disease genes and
candidate genes in patients with CTD is a focus of current
cardiovascular research. Studies in animal models have demonstrated
that the GATA binding protein 6 (GATA6) regulates
differentiation and affects the development of CTDs. Homozygous
GATA6-knockout mice exhibit developmental endoderm defects
that lead to embryonic death (7,8).
Studies have demonstrated that conditional inactivation of
GATA6 in vascular smooth muscle cells (VSMCs) in mice
results in perinatal mortality from a spectrum of cardiovascular
defects, including IAA and PTA. Inactivation of GATA6 in the
neural crest recapitulates these abnormalities, demonstrating a
cell-autonomous requirement for GATA6 in neural
crest-derived smooth muscle cells (9). It is therefore necessary to screen
for GATA6 mutations in patients with nonsyndromic
conotruncal heart defects to enable early disease intervention and
genetic counseling.
Materials and methods
Subjects
The present study was approved by the Medical Ethics
Committee of Xinhua Hospital (Shanghai, China). Once informed
consent was obtained from the parents of all patients and control
subjects, venous blood samples from all participants were collected
in an anticoagulant tube with sodium citrate. Karyotype analysis
and multiplex ligation-dependent probe amplification were performed
in samples from all patients with CTDs. Fluorescence in situ
hybridization (FISH) was performed in all patients to identify and
exclude patients with genetic deletions such as trisomy 18, trisomy
21 and 22q11.2 deletion. Nonsyndromic patients with CTD were
diagnosed by transthoracic echocardiography, computed tomography,
cardiac catheterization, and/or surgical inspection. A total of 157
unrelated Chinese patients with nonsyndromic CTD were enrolled in
the study from January 2009 to January 2011. Patients included 105
males and 52 females, aged between 1 month and 17 years old with a
median age of 3.64 years. CTDs in these patients included: TOF (73
cases); PA/VSD (27); DORV
(28); TGA (9); PTA(10); and IAA (10). The present study enrolled 300
healthy unrelated children as healthy controls. All participants
were of Han ethnicity. Genomic DNA was isolated from 200 μl blood
using a standard phenol-chloroform extraction protocol. The
families of the probands (parents and siblings) also underwent
physical examination and transthoracic echocardiography, and venous
blood samples were analyzed for GATA6 mutations.
Screening for mutations
Seven whole exons and exon-intron boundaries of
human GATA6 were amplified. Oligonucleotide primers were
designed based on genomic sequences (GenBank accession number
NC_000018) using Primer3 software (http://frodo.wi.mit.edu/primer3/) and were synthesized
by Shanghai Genesky Bio-Tech (Shanghai, China). Primers were
designed so that each exon was flanked by part of the corresponding
intron (Table I). For exons 1, 2,
3 and 7, the polymerase chain reaction (PCR) reaction mixture
(total, 10 μl) contained 1.0 μl genomic DNA, 1.0 μl each primer,
0.2 μl dNTP mixture, 5.0 μl 2× GC buffer I, 2.74 μl
ddH20, and 0.06 μl HotTaq DNA polymerase (Takara
Biotechnology, Dalian, China). PCR was performed using a GeneAmp
9600 Thermal cycler (Applied Biosystems, Foster City, CA, USA)
under the following conditions: Predenaturation at 95°C for 2 min,
followed by 35 cycles at 96°C for 10 sec, and annealing and
extension at 72°C for 4 min. For exons 4 and 5–6, the PCR reaction
mixture (total, 10 μl) contained 1.0 μl genomic DNA, 1.0 μl each
primer, 0.2 μl dNTP mixture, 1.0 μl 2× GC buffer I, 0.2 μl
MgC12, 6.54 μl ddH20, and 0.06 μl of HotTaq
DNA polymerase. PCR cycling conditions were as follows: 11 cycles
of predenaturation at 95°C for 2 min, denaturation at 96°C for 20
sec, denaturation at 62°C for 40 sec, and extension at 72°C for 2
min (annealing temperature was decreased by 0.5°C/cycle); 24 cycles
of denaturation at 94°C for 20 sec, annealing at 56°C for 30 sec
and extension at 72°C for 2 min. All PCR products were gel purified
using a QIAquick Gel Extraction kit (Qiagen, Hilden, Germany) and
then sequenced using the dideoxy chain termination method on an
ABI3130XL sequencer (Applied Biosystems). Sequencing results were
aligned with the reference sequence using the GenBank BLAST program
(http://blast.ncbi.nlm.nih.gov/Blast.cgi). The
corresponding sequences of healthy controls were amplified and
sequenced as above in order to exclude polymorphisms. GATA6
protein sequences from various species were aligned using ClustalW
software (www.clustal.org).
 | Table IPrimers used to amplify the
GATA6 gene. |
Table I
Primers used to amplify the
GATA6 gene.
| Fragment | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Fragment size
(bp) |
|---|
| 1 |
ccgtcccctccccaccctcttt |
gagatcgcgcgcgaggaggaagca | 361 |
| 2–1 |
tggaggcgaggtagcgtgcag |
ctcgggtgcgaaggggctcag | 544 |
| 2–2 |
cccgctcgctgctgctcagtt |
ccatgggcgggctgggagagt | 591 |
| 2–3 |
cacctgcaggggtcgggcagt |
aaacagggcccgagtggagca | 616 |
| 3 |
ctactggggcgctccgggtgt |
agcgggtgggcgttggaacag | 583 |
| 4 |
tggagaagaaaccagggatga |
tgcattcaaatttttcacttgag | 590 |
| 5–6 |
cggcggccaaattctttta |
aaccataaaaaaatgataccgatct | 619 |
| 7 |
tggccagggtcaggtcagtgg |
gagtggcccaagcgcccagtt | 610 |
Plasmid construction and cells
The wild-type GATA6 expression plasmid,
pcDNA3.1(+)-Homo GATA6 was supplied by Professor Hiroyuki
Yamagishi (10). To generate the
GATA6 E51K and G245R mutant constructs, mutations were
introduced into pcDNA3.1(+)-Homo GATA6 by site-directed
mutagenesis-PCR individually. The vector with a luciferase
reporter driven by the ANF promoter was a kind gift from Professor
Vidu Garg (11). HEK293T cells
(Cell Bank of the Chinese Academy of Science, Shanghai, China) were
split and seeded into 96-well plates with 10,000 cells/well in
preparation for the following assay.
Transfection and transcriptional
assay
Transfection using FuGene HD Transfection reagent
(Roche Diagnostics, Mannheim, Germany) was conducted in triplicate
as previously described (12), and
then the activity of firefly luciferase and LacZ in cell
lysates was measured by the Dual-Glo Luciferase Assay system
(Promega Corporation, Madison, WI, USA) 24 h after transfection.
HEK239T cells were transfected with 20 ng of wild-type or mutant
GATA6, 100 ng reporter construct ANF-luciferase, and 20 ng
cytomegalovirus (CMV)-LacZ in each well for correcting the
transfection efficiency. Results are presented as the relative
luciferase activity, normalized to the co-transfected
CMV-LacZ group.
Statistical analysis
Data are presented as the mean ± standard deviation.
The two groups were compared using the χ2 test for
continuous variables and P<0.05 was considered to indicate a
statistically significant difference.
Results
GATA6 mutations in patients with
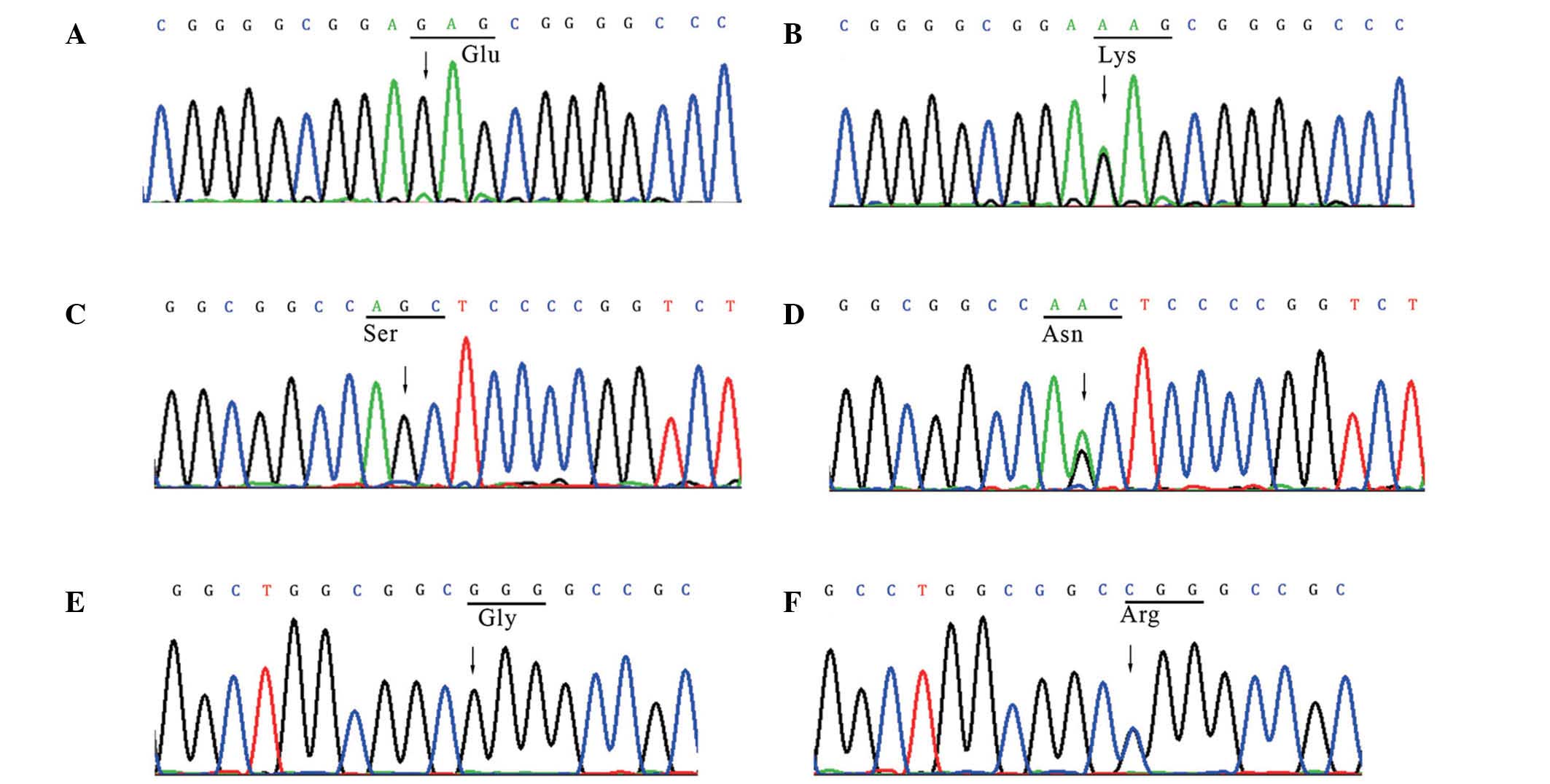
nonsyndromic CTD
Three heterozygous missense mutations (E51K, S184N
and G245R) were identified in the GATA6 gene in 3/157
unrelated cases of nonsyndromic CTD. These sequence variants, which
are located in coding exons and generate the amino acid alterations
of E51K, S184N and G245R, were absent in the chromosomes of all
healthy controls (Fig. 1).
Overall, the mutation frequency of the GATA6 gene in
nonsyndromic CTD was 1.9% (3/157). To the best of our knowledge,
the current study is the first to report the E51K and G245R
heterozygous missense mutations. The c.151G>A in exon 2,
identified in a patient with TOF, changes a relatively conserved
glutamic acid residue to lysine at position 51 (E51K). This change
was expected to have a significant impact on the structure and
function of the GATA6 protein since glutamic acid is acidic,
while lysine is basic. The c.733G>C missense mutation in exon 2,
identified in a patient with PTA, leads to the substitution of a
highly conserved glycine residue with arginine at position 245
(G245R). This mutation may also have a significant impact on the
structure of a salt bond and thus the structure of the GATA6
protein since glycine is a non-polar hydrophobic amino acid, while
arginine is basic. In addition, the c.551G>A missense mutation
in exon 2 was identified in a patient with TOF, and substitutes a
serine with an asparagine at position 184 (S184N). However, this
mutation has been reported previously (13). As the two novel mutations are
likely to alter the structure of the GATA6 protein, they may
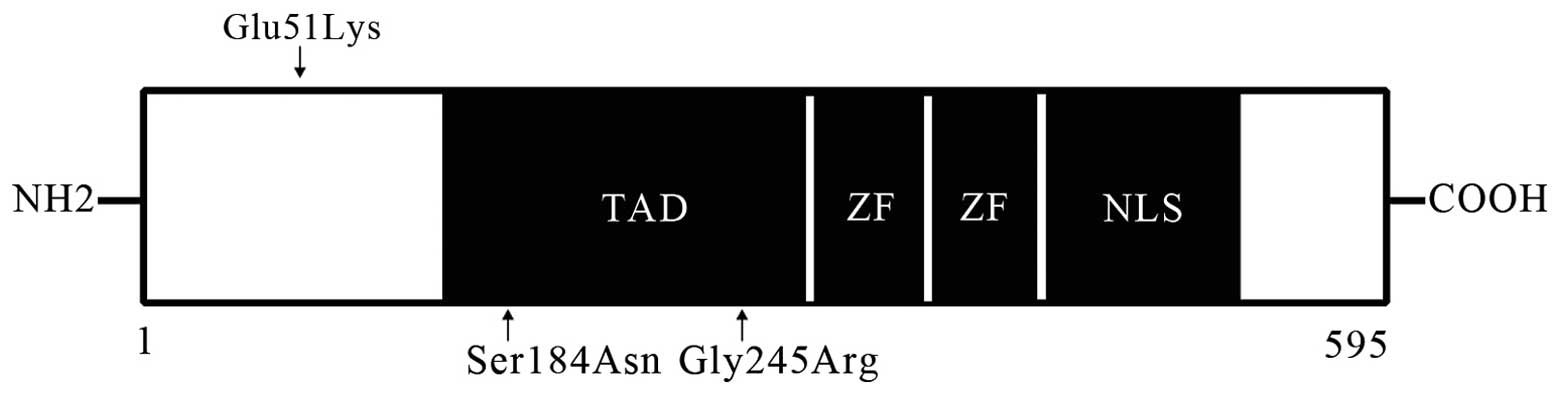
be pathogenic. The location and sequence alignment data for these
mutations are shown in Figs. 2 and
3. Nonsyndromic CTD pathogenesis
is not only related with the 22q11.2 microdeletion and TBX1
gene mutations, but may also be involved in pathogenesis mediated
by the GATA6 zinc-finger transcription factor.
Seven sequence variants were identified in introns
and the 3′-UTR of GATA6 in patients with nonsyndromic CTD
and control subjects. A previously reported sequence variant in
exon 2 (c.43G>C) predicted to lead to an amino acid change
(p.Gly15Arg) was also identified in the present study. This
nucleotide substitution was identified in 8/157 nonsyndromic CTD
patients (5.1%) and in 21/300 control subjects (7.0%) (P>0.05).
Six of these sequence variants were already present in the dbSNP
database (http://www.ncbi.nlm.nih.gov/SNP/). The other two
intron sequence variants (c.10501-85T>C, intron 2;
c.16589+6T>C, intron 6) were novel single nucleotide
polymorphisms, but displayed no significant difference in their
allele frequencies between patients with nonsyndromic CTD and
control subjects. All sequence variants and their allele
frequencies are summarized in Table
II.
| Table IIIdentification of GATA6
sequence variants in nonsyndromic CTD. |
Table II
Identification of GATA6
sequence variants in nonsyndromic CTD.
| | | | Allele
frequency |
|---|
| | | |
|
|---|
| Location | Nucleotide
change | dbSNP database | Amino acid
change | Patients | Controls |
|---|
| Exon 2 | c.43G>C | rs116262672 | Gly15Arg | 8/157
(0.051)a | 21/300 (0.070) |
| Exon 2 | c.151G>A | | Glu51Lys | 1/157
(0.006)b | 0/300 (0.000) |
| Exon 2 | c.551G>A | | Ser184Asn | 1/157
(0.006)b | 0/300 (0.000) |
| Exon 2 | c.733G>C | | Gly245Arg | 1/157
(0.006)b | 0/300 (0.000) |
| Intron 2 |
c.5825+19C>G | rs76308670 | | 1/157
(0.006)a | 2/300 (0.007) |
| Intron 2 |
c.10501-60C>T | rs3764504 | | 4/156
(0.026)a | 8/300 (0.027) |
| Intron 2 |
c.10501-85T>C | | | 1/157
(0.006)a | 2/300 (0.006) |
| Intron 6 |
c.16589+6T>C | | | 1/157
(0.003)a | 2/300 (0.007) |
| Intron 6 |
c.16589+7A>G | rs3764962 | | 9/157
(0.057)a | 22/300 (0.073) |
| 3′-UTR | c.+72G>A | rs1941084 | | 67/157
(0.427)a | 156/300
(0.520) |
| 3′-UTR | c.+77G>A | rs1941083 | | 26/157
(0.166)a | 56/300 (0.187) |
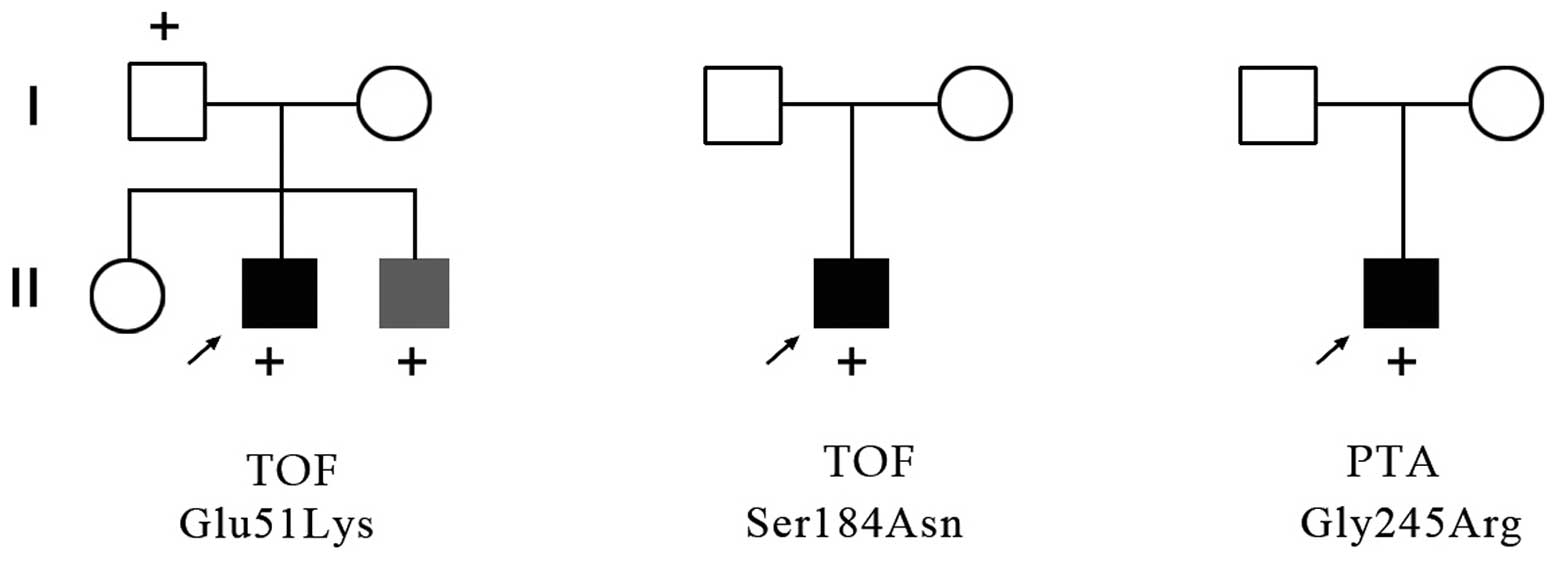
Characteristics and mutation screening of
family members
The c.151G>A proband was a male with TOF, and
family members included two parents and siblings. A young brother
of the proband was confirmed to be suffering from trisomy 21
associated with an atrioventricular septal defect (AVSD), but his
parents and sister had normal cardiac morphology. The c.151G>A
missense mutation was identified in his father and brother. The
c.551G>A and the c.733G>C probands were males with TOF and
PTA, respectively, and family members included parents but no
siblings in both cases. These parents all had normal cardiac
morphology and there were no GATA6 sequence variants
identified (Fig. 4).
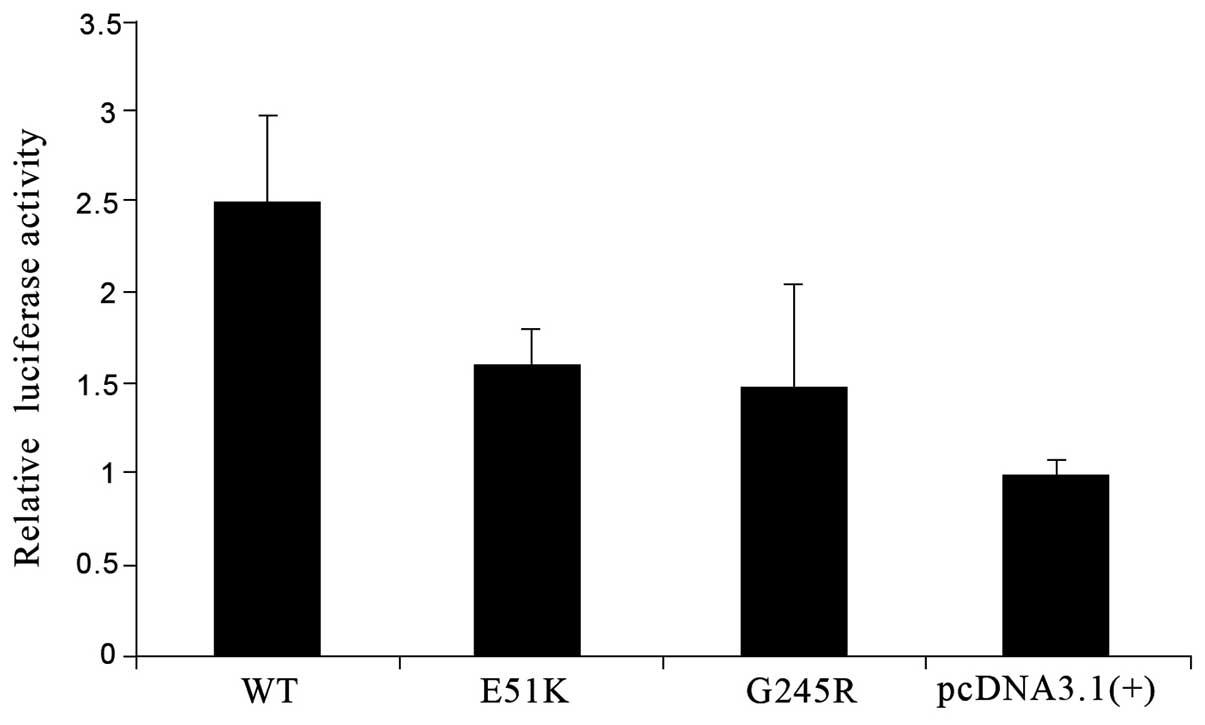
E51K and G245R cause a significant
reduction in the capacity of GATA6 to transactivate downstream
genes
Among the three mutations (E51K, S184N and G245R),
S184N has been reported in a previous study by Lin et al
(13). In the current study, the
transcription capacities of the E51K and G245R proteins in cultured
cells were determined. The ANF-luciferase construct in which the
reporter gene expression was driven by the ANF promoter was
co-transfected into HEK293T cells with wild-type or mutant
GATA6. The expressed GATA6 molecules bound to the ANF
sequence and then transactivated luciferase expression downstream.
The luciferase activity normalized to the LacZ value was
regarded as the relative transactivation function of GATA6.
E51K diminished the transactivation activity by almost 40% and
G245R interrupted it more markedly (>40%) compared with that of
wild-type GATA6 (Fig.
5).
Discussion
GATA6 is an early marker of cardiac precursor
cells, an important transcription factor for cardiac cell
differentiation and development, and one of the major candidate
genes in the pathogenesis of congenital heart diseases,
particularly conotruncal heart defects (10,14).
The human GATA6 gene is localized to chromosome
18q11.1–11.2, contains seven exons spanning 34,812 bp and encodes a
protein comprising 595 amino acids (15). GATA6 expression in the
cardiac neural crest during embryonic development can be visualized
with FISH, and data obtained using this technique supports the
hypothesis that GATA6 not only regulates differentiation of
smooth muscle cells in the cardiac neural crest but also regulates
development of the aorticopulmonary septum through signaling from
cells derived from the neural crest (9,16).
In the wild-type embryonic heart, GATA6 mRNA is expressed in
the cardiac outflow tract, atrium and ventricles 9.5 days
post-conception. GATA6 is expressed in the ascending aorta,
pulmonary artery smooth muscle cells, and the endocardial cushion
that differentiates into the cardiac outflow tract conotruncus 11.5
days post-conception. Furthermore, GATA6 mRNA detected at
12.5 days post-conception is localized in the aorta, pulmonary
artery, VSMCs of the ductus arteriosus, cardiomyocytes of the
atrium and ventricles, and the endocardial cushion of the
conotruncus (17,18). It is reported that of the GATA gene
family, only GATA6 is expressed in VSMCs and it mediates
their differentiation through modulating the expression of
VSMC-specific genes, including α-MHC, α-actin and SM22, in
order to maintain various differentiated vascular smooth muscle
phenotypes (19–23).
Three GATA6 missense mutations that were
demonstrated to be associated with single conotruncal heart defects
were identified in the current study, and two of these were novel
(E51K and G245R). The E51K mutation was not demonstrated to be
located in the transcriptional activation, zinc finger or nuclear
localization signaling domains; however, this mutation results in
the replacement of an acidic with a basic amino acid at position
51. Furthermore, the region of the GATA6 sequence containing
this mutation is important in the transactivation of target genes.
It is therefore reasonable to suggest that the E51K mutation
significantly affects the structure, stability, and hence, the
transcriptional activity of the GATA6 protein. It was also
indicated that the G245R mutation results in changes to salt bonds
and may alter the structure of the GATA6 protein.
These assumptions were confirmed by functional
analysis in the present study. The E51K and G245R mutations reduced
the capacity of GATA6 to transactivate downstream genes by
~40% compared with that of wild-type GATA6. Lin et al
(13) previously identified S184N
missense mutations in one patient with TOF and one with an atrial
septal defect, and then demonstrated that the mutation led to
reduced transcriptional activity of GATA6 (13). The mutant GATA6 appears to
attenuate the expression of certain downstream genes which modulate
the formation of the cardiac outflow tract, endocardial cushion and
the atrioventricular septa, and this may be the genetic mechanism
underlying nonsyndromic CTDs.
Thus far, studies have reported GATA6
mutations in congenital heart disease. Kodo et al (10) reported the N466H mutation in the
zinc finger domain and the E486del mutation in the nuclear
localization signaling domain in patients with PTA. They also
demonstrated that the mutant proteins did not transactivate target
genes, thus disrupting normal regulation of the semaphorin-plexin
signaling pathway and resulting in cardiac outflow tract
malformations. Maitra et al (24) identified two missense mutations of
the GATA6 gene in one patient with TOF (A178V) and one with
AVSD (L198V). They also demonstrated that the A178V mutation leads
to increased GATA6 transcriptional activity. In the present
study, a detailed phenotype and genotype characterization of the
family members of all patients with GATA6 mutations was
performed. The results displayed that the father and brother of the
proband with the E51K mutation also had the mutation (c.151G>A);
however, the father had normal cardiac morphology, suggesting
incomplete penetrance of this mutation. The brother was diagnosed
with trisomy 21 syndrome with AVSD. It is reported that ~40–60% of
patients with trisomy 21 syndrome exhibit some form of congenital
heart disease; however, trisomy 21 alone is insufficient to cause
congenital heart disease as ~50% of patients with trisomy 21 have a
normal heart (25,26). It has been suggested that the
etiology for heart defects in trisomy 21 with AVSD involves
mutations in the CRELD1, HEY2 and ALK2 genes
(26–28). The present study suggests that
mutation of the GATA6 gene may also be involved in the
pathogenesis of AVSD.
The results of the present study demonstrated that
mutations of the GATA6 gene are closely related to
nonsyndromic CTDs, suggesting an important role for this gene in
the development of the human heart conotruncus. However, the
development and differentiation of the human heart is a complex
process and many factors contribute to the pathogenesis of abnormal
heart structures, including genetic mutation, epigenetic
modification and abnormal gene-environment interactions. Further
studies are therefore required to clarify the role of the
GATA6 mutations in the pathogenesis of heart malformations,
particularly in CTD.
Acknowledgements
The authors thank all the patients and their
families who participated in this study, Professor Hiroyuki
Yamagishi and Professor Vidu Garg for kindly offering plasmids, and
all the staff of the Children’s Heart Center of Xinhua Hospital for
their assistance. This study was supported by the ‘973’ Program
Fund of China (2010CB529501), the funding of Shanghai sample of
clinical research center construction projects (08DZ2293105) and
the Shanghai University Innovation Team.
References
|
1
|
Hoffman JI and Kaplan S: The incidence of
congenital heart disease. J Am Coll Cardiol. 39:1890–1900. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Debrus S, Berger G, de Meeus A, et al:
Familial non-syndromic conotruncal defects are not associated with
a 22q11 microdeletion. Hum Genet. 97:138–144. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jenkins KJ, Correa A, Feinstein JA, et al:
The American Heart Association Council on Cardiovascular Disease in
the Young: Noninherited risk factors and congenital cardiovascular
defects: current knowledge: a scientific statement from the
American Heart Association Council on Cardiovascular Disease in the
Young: endorsed by the American Academy of Pediatrics. Circulation.
115:2995–3014. 2007.
|
|
4
|
Cooper WO, Hernandez-Diaz S, Arbogast PG,
et al: Major congenital malformations after first-trimester
exposure to ACE inhibitors. N Engl J Med. 354:2443–2451. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goldmuntz E, Clark BJ, Mitchell LE, et al:
Frequency of 22q11 deletions in patients with conotruncal defects.
J Am Coll Cardiol. 32:492–498. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu YJ, Wang J, Xu R, et al: Detecting
22q11.2 deletion in Chinese children with conotruncal heart defects
and single nucleotide polymorphisms in the haploid TBX1 locus. BMC
Med Genet. 12:1692011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morrisey EE, Tang Z, Sigrist K, et al:
GATA6 regulates HNF4 and is required for differentiation of
visceral endoderm in the mouse embryo. Genes Dev. 12:3579–3590.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao R, Watt AJ, Battle MA, Li J, Bondow
BJ and Duncan SA: Loss of both GATA4 and GATA6 blocks cardiac
myocyte differentiation and results in acardia in mice. Dev Biol.
317:614–619. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lepore JJ, Mericko PA, Cheng L, Lu MM,
Morrisey EE and Parmacek MS: GATA-6 regulates semaphorin 3C and is
required in cardiac neural crest for cardiovascular morphogenesis.
J Clin Invest. 116:929–939. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kodo K, Nishizawa T, Furutani M, et al:
GATA6 mutations cause human cardiac outflow tract defects by
disrupting semaphorin-plexin signaling. Proc Natl Acad Sci USA.
106:13933–13938. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sprenkle AB, Murray SF and Glembotski CC:
Involvement of multiple cis elements in basal- and alpha-adrenergic
agonist-inducible atrial natriuretic factor transcription. Roles
for serum response elements and an SP-1-like element. Circ Res.
77:1060–1069. 1995. View Article : Google Scholar
|
|
12
|
Bamforth SD, Bragança J, Eloranta JJ, et
al: Cardiac malformations, adrenal agenesis, neural crest defects
and exencephaly in mice lacking Cited2, a new Tfap2 co-activator.
Nat Genet. 29:469–474. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin X, Huo Z, Liu X, et al: A novel GATA6
mutation in patients with tetralogy of Fallot or atrial septal
defect. J Hum Genet. 55:662–667. 2010. View Article : Google Scholar
|
|
14
|
Burch JB: Regulation of GATA gene
expression during vertebrate development. Semin Cell Dev Biol.
16:71–81. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brewer A and Pizzey J: GATA factors in
vertebrate heart development and disease. Expert Rev Mol Med.
8:1–20. 2006. View Article : Google Scholar
|
|
16
|
Barillot W, Tréguer K, Faucheux C, Fédou
S, Thézé N and Thiébaud P: Induction and modulation of smooth
muscle differentiation in Xenopus embryonic cells. Dev Dyn.
237:3373–3386. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stoller JZ and Epstein JA: Cardiac neural
crest. Semin Cell Dev Biol. 16:704–715. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brown CB, Feiner L, Lu MM, et al: PlexinA2
and semaphorin signaling during cardiac neural crest development.
Development. 128:3071–3080. 2001.PubMed/NCBI
|
|
19
|
Yang H, Lu MM, Zhang L, Whitsett JA and
Morrisey EE: GATA6 regulates differentiation of distal lung
epithelium. Development. 129:2233–2246. 2002.PubMed/NCBI
|
|
20
|
Fischer A, Klattig J, Kneitz B, et al: Hey
basic helix-loop-helix transcription factors are repressors of
GATA4 and GATA6 and restrict expression of the GATA target gene ANF
in fetal hearts. Mol Cell Biol. 25:8960–8970. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suzuki E, Evans T, Lowry J, Truong L, Bell
DW, Testa JR and Walsh K: The human GATA-6 gene: structure,
chromosomal location, and regulation of expression by
tissue-specific and mitogen-responsive signals. Genomics.
38:283–290. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mano T, Luo Z, Malendowicz SL, Evans T and
Walsh K: Reversal of GATA-6 downregulation promotes smooth muscle
differentiation and inhibits intimal hyperplasia in balloon-injured
rat carotid artery. Circ Res. 84:647–654. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morrisey EE: GATA-6: the proliferation
stops here: cell proliferation in glomerular mesangial and vascular
smooth muscle cells. Circ Res. 87:638–640. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maitra M, Koenig SN, Srivastava D and Garg
V: Identification of GATA6 sequence variants in patients with
congenital heart defects. Pediatr Res. 68:281–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Loffredo CA, Hirata J, Wilson PD, Ferencz
C and Lurie IW: Atrioventricular septal defects: possible etiologic
differences between complete and partial defects. Teratology.
63:87–93. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li H, Cherry S, Klinedinst D, et al:
Genetic modifiers predisposing to congenital heart disease in the
sensitized Down syndrome population. Circ Cardiovasc Genet.
5:301–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guo Y, Shen J, Yuan L, Li F, Wang J and
Sun K: Novel CRELD1 gene mutations in patients with
atrioventricular septal defect. World J Pediatr. 6:348–352. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Joziasse IC, Smith KA, Chocron S, et al:
ALK2 mutation in a patient with Down’s syndrome and a congenital
heart defect. Eur J Hum Genet. 19:389–393. 2011.PubMed/NCBI
|