Introduction
Opioids, such as morphine, have been widely utilized
clinically as one of the most potent analgesics for treating acute
and chronic pain conditions. However, the clinical utility of
opioid analgesics is often limited by the development of analgesic
tolerance, that necessitates dose escalation. An increasing number
of studies are investigating the neurobiological underpinnings of
opioid tolerance, including those on N-methyl-D-aspartate (NMDA)
receptors (1,2), proinflammatory cytokines
[interleukin-1β (IL-1β), IL-6 and tumor necrosis factor-α (TNF-α)]
(3,4) and signal transducer and activator of
transcription 3 (STAT3) (5).
Recently, we have demonstrated the role of p38 mitogen-activated
protein kinase (MAPK) in the development of morphine
antinociceptive tolerance (6,7).
Based on the data from these studies, it was proposed that the
molecules, which can modulate elements such as NMDA receptors,
IL-1β, p38 MAPK and STAT3, may contribute to the development of
morphine antinociceptive tolerance. It was hypothesized that one
candidate molecule for this may be leptin.
Leptin, an adipokine, is a 16 kDa, nonglycosylated
peptide hormone encoded by the obese gene (ob) in mice (8). Stimulation of its receptor leads to
activation of the Janus kinase (JAK)-STAT signaling pathway
(9). Numerous studies have
revealed that leptin is crucial in nociceptive behavior elicited by
nerve injury in rats (10,11). While the peripheral effect of
leptin on neuropathic pain is mediated by macrophage stimulation
(10), its central effect is
likely associated with the upregulation of NMDA receptors following
nerve injury (11). A more recent
study also reported that intrathecal (i.t.) leptin enhances the
expression of NMDA receptors and phosphorylated (p)-STAT3 within
the rat spinal cord dorsal horn (12). Since it was hypothesized that
morphine tolerance and neuropathic pain share certain common
pathological mechanisms (13,14),
the present study investigated the hypothesis that (i) spinal
leptin would be involved in the development of morphine
antinociceptive tolerance in rats and (ii) the proposed spinal
leptin effect would be mediated by leptin-dependent downstream
cellular responses, including the increased expression of spinal
STAT3 and NMDA receptors. The results demonstrated that chronic
i.t. administration of morphine significantly upregulates the
expression levels of spinal leptin, leptin receptor (Ob-R) and
p-STAT3, as well as the NR1 subunit of NMDA receptors in the spinal
cord. The data suggest that the activation of spinal STAT3-NMDA
receptor pathway contributes to leptin-mediated development of
morphine antinociceptive tolerance in rats.
Materials and methods
Materials
Morphine hydrochloride was purchased from Qinghai
Pharmaceutical Factory Co., Ltd. (Xining, Qinghai, China). Leptin
antagonist (LA) was obtained from ProSpec-Tany TechnoGene Ltd.
(East Brunswick, NJ, USA). AG490 and MK-801 were purchased from
Sigma-Aldrich (St. Louis, MO, USA).
Animals
Adult male Sprague-Dawley rats (weight, 250–280 g)
were provided by Sun Yat-sen University Experimental Animal Center
(Guangzhou, Guangdong, China) and housed individually with free
access to water and food at a temperature of 22±2°C and in a light
control (12 h light-dark cycle) quiet room. All experimental
procedures were approved by the ethics committee of Zhongshan
School of Medicine, Sun Yat-sen University (Guangzhou, China) and
were conducted strictly in compliance with the NIH guidelines for
the Care and Use of Laboratory Animals.
I.t. catheter implantation
For i.t. delivery of the indicated drugs (such as
morphine, etc.), rats were implanted with i.t. catheters according
to the methods described previously (6). Briefly, a sterile polyethylene
(PE-10) tube (length=7 cm) filled with saline was inserted through
the L5/L6 intervertebral space and the tip of the tube was placed
at the spinal lumbar enlargement level. Correct i.t. catheter
placement was verified following the completion of behavioral test
by visual inspection. The wound was closed in two layers with 4-0
polyester suture. All the animals were allowed to recover for at
least 4 days prior to the experiments. Any rats that developed hind
limb paralysis or paresis following surgery were excluded and
euthanized with an overdose of pentobarbital.
Drug administration
Drugs were administered in volumes of 10 μl followed
by a flush of 7 μl of saline to ensure drug delivery into the
subarachnoid space via the i.t. catheter. Morphine hydrochloride
was dissolved in saline. According to the previously described
methods, LA (3 μg) (11), AG490 (1
μg) (11) and MK-801 (10 nmol)
(12) were dissolved in DMSO and
diluted by saline to a final volume of 10 μl. The residual drug
solution was discarded following each experiment.
Induction of morphine antinociceptive
tolerance and behavioral test
Tolerance to morphine antinociceptive effect was
induced by i.t. administration of morphine (15 μg daily) for 7
days. Nociceptive tests were performed prior to and 30 min
following morphine administration on day 1, 3, 5 and 7. To examine
the effect of LA, AG490 or MK-801 on morphine antinociceptive
tolerance, drugs were administered via i.t. 30 min prior to each
morphine administration, respectively.
Morphine antinociception was evaluated by the
hot-water tail-flick test according to the previously described
methods (6). Briefly, rats were
restrained in a plastic container (22×6 cm) and the distal third of
the tail was immersed into the water maintained at 50±0.2°C. The
latency response was defined by rapid removal of the tail from the
water. A cut off time of 15 sec was used to avoid tissue damage.
Three trials were performed for each rat with an intertrial
interval of 2 min and the mean of three measurements was obtained
as the final latency. The percentage of maximal possible
antinociceptive effect (%MPE) was calculated by comparing the test
latency prior to [baseline (BL)] and post-drug injection (TL) using
the equation: %MPE = [(TL-BL)/(cut off time-BL)] × 100%.
Immunohistochemistry
Immediately following the behavioral test on the
indicated days, rats were deeply anesthetized with sodium
pentobarbital (100 mg/kg, i.p.), and perfused through the ascending
aorta with cold saline, followed by 4% paraformaldehyde in 0.1 M
PBS (pH 7.2–7.4, 4°C). Following perfusion, the spinal cord lumbar
enlargement was quickly removed and post-fixed in the same fixative
for 2 h and then cytoprotected in 30% sucrose for two nights.
Transverse spinal sections (20 μm) were cut in a cryostat, mounted
on polylysine-coated slides and processed for immunohistochemistry.
All of the sections were blocked with 2% goat or donkey serum in
0.1 M TBS/0.3% Triton X-100 for 1 h at room temperature (RT) and
incubated over two nights at 4°C with the primary antibody for
leptin (Ob; dilution, 1:100), leptin receptor (Ob-R; dilution,
1:50; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) or
p-STAT3 (Tyr705; dilution, 1:50; Cell Signaling Technology, Inc.,
Danvers, MA, USA), NMDA receptor NR1 subunit (NR1; dilution, 1:100;
Bioworld Technology, Inc., MN, USA), glial fibrillary acidic
protein (GFAP; astrocyte marker; dilution, 1:400), OX-42 (microglia
marker; dilution, 1:400) or NeuN (neuron marker; dilution, 1:300;
Merck Millipore, Billerica, MA, USA). For double immunofluorescent
staining, the primary antibody for the protein of interest and the
marker for the above cells were mixed. Then, the slides were
incubated away from the light at RT for 2 h with the corresponding
single or mixed FITC- or Cy3-conjugated secondary fluorescent
antibody (dilution, 1:400; Jackson ImmunoResearch, Western Grove,
PA, USA). The stained sections were observed with an Olympus
fluorescence microscope (Olympus, Tokyo, Japan) and the images were
captured by a CCD spot Camera. Non-specific staining was determined
by omitting the primary antibodies.
Western blot assay
The lumbar enlargements of the spinal cord were
removed immediately following the behavioral tests and placed into
liquid nitrogen for quick freezing. The tissue was put into
radioimmunoprecipitation assay (RIPA) buffer containing protease
and phosphatase inhibitor cocktails (Sigma-Aldrich) to be
homogenized on ice. Following centrifugation at 4°C, the
supernatants were collected. Then, the protein samples were
separated by SDS-polyacrylamide gel electrophoresis and
electrotransferred onto polyvinylidene fluoride membrane (Merck
Millipore). The membrane was blocked by 5% non-fat milk for 1 h at
RT, then incubated overnight at 4°C with the primary antibody for
leptin (Ob; dilution, 1:1,000) or leptin receptor (Ob-R; dilution,
1:500, Santa Cruz Biotechnology, Inc.) or p-STAT3 (Tyr705;
dilution, 1:500; Cell Signaling Technology, Inc.) or NMDA receptor
NR1 (NR1; dilution, 1:500; Bioworld Technology, Inc.),
respectively. Then, the membrane was incubated with the horseradish
peroxidase labeled secondary antibody (1:3,000; Bioworld
Technology, Inc.) for 1.5 h at RT, developed in ECL solution and
exposed onto X-ray film.
Statistical analysis
All data are presented as the mean ± SEM.
Differences between groups were analyzed by one-way analysis of
variance (ANOVA) using SPSS 17.0 software and followed by LSD post
hoc comparison test. P<0.05 was considered to indicate a
statistically significant result.
Results
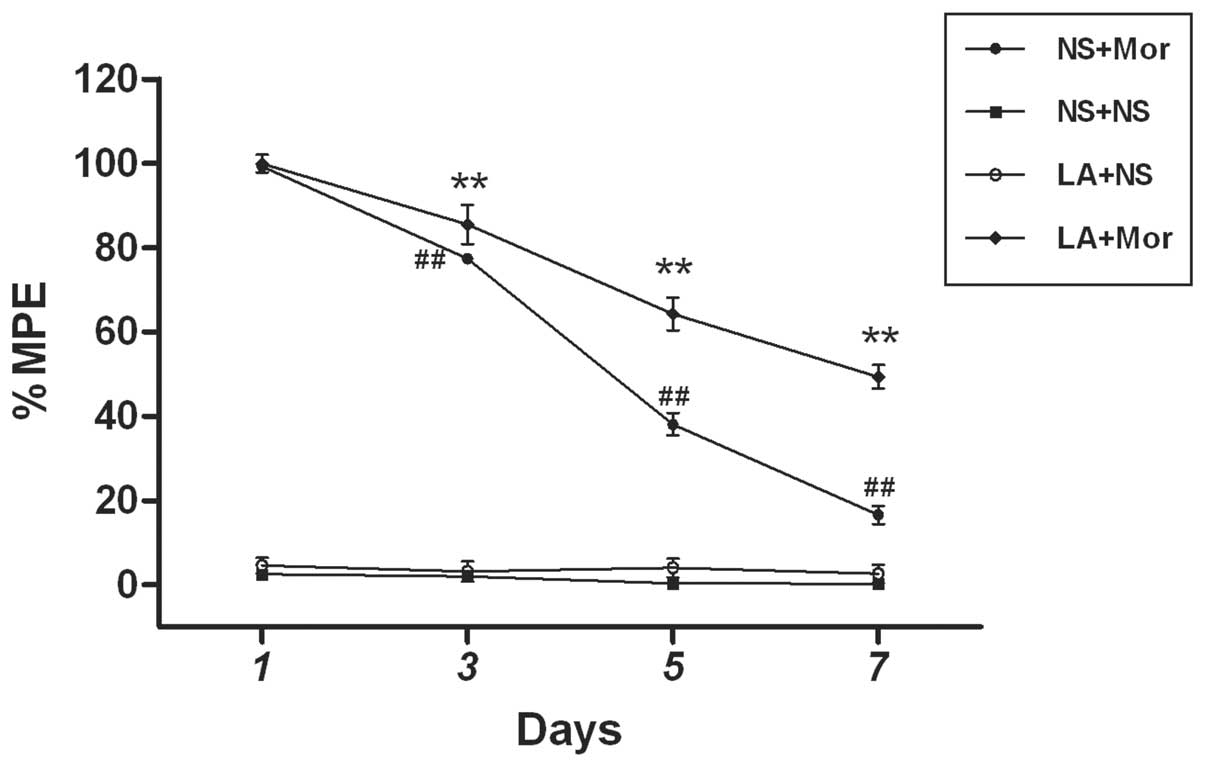
Spinal leptin critically contributes to
morphine antinociceptive tolerance in rats
To test whether the blockage of spinal leptin would
prevent the development of morphine antinociceptive tolerance
following chronic morphine exposure, a LA was administered via i.t.
once daily for 7 days, 30 min prior to morphine treatment. This
treatment regimen significantly reduced the development of morphine
antinociceptive tolerance when examined at 30 min following
morphine treatment on days 3, 5 and 7, as compared with the
morphine treatment group (Fig. 1).
However, repeated exposure to normal saline (NS) or LA (3 μg) alone
did not elicit any analgesic effect as compared with the saline
group assessed by the tail-flick test (Fig. 1). These results suggested that the
spinal leptin is critical in the development of morphine
antinociceptive tolerance.
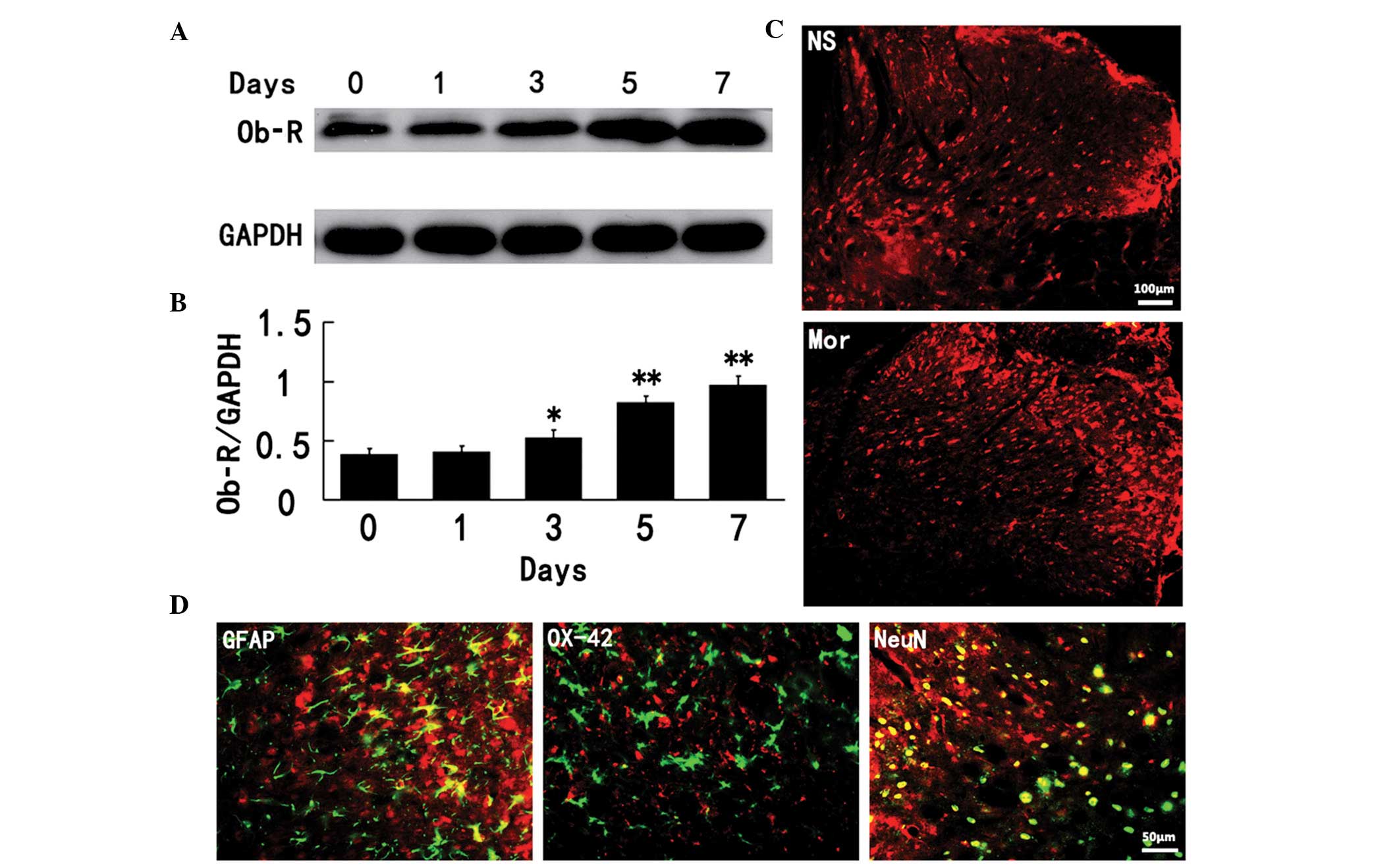
Chronic morphine exposure induces the
upregulation of spinal leptin receptor expression in rats
To examine the molecular mechanisms responsible for
the spinal leptin effects on morphine antinociceptive tolerance, we
first determined whether the expression of leptin receptor (Ob-R)
in the spinal cord would be altered following chronic morphine
treatment because Ob-R is considered to be responsible for the
majority of leptin functions (15,16,17).
As demonstrated by the data of the western blot assay, the
expression of spinal Ob-R protein was enhanced at a time course
similar to that of the development of morphine antinociceptive
tolerance (Fig. 2A and B). In
addition, Ob-R immunoreactivity was topographically increased in
the spinal cord dorsal horn of chronic morphine treatment rats
(Fig. 2C). The spinal Ob-R
immunoreactivity was colocalized primarily with NeuN (a neuronal
marker) and, to a lesser extent, with GFAP (an astrocyte marker;
Fig. 2D), revealing that chronic
morphine exposure mainly induced the upregulation of neuronal Ob-R
in the spinal cord dorsal horn of rats.
Chronic morphine exposure enhances the
spinal expression of leptin
The changes in spinal leptin expression during the
development of morphine antinociceptive tolerance were examined. As
illustrated in Fig. 3, the
increased spinal leptin expression (western blot analysis) on day 1
following morphine treatment was not statistically significant as
compared with the saline group. By contrast, the increases in
spinal leptin expression observed on days 3, 5 and 7, were
significantly different in the morphine-treated rats, as compared
with the control group (P<0.05), respectively, as evidenced by a
time-dependent increase in the spinal leptin expression following
chronic morphine exposure.
LA reduces chronic morphine-induced
phosphorylation of the spinal STAT3
Thirdly, we examined whether there was a correlation
between the spinal STAT3 activation and leptin, following chronic
exposure to morphine. The data from the western blot analysis
demonstrated that the phosphorylated expression of spinal STAT3 was
markedly increased on day 7 following chronic morphine treatment as
compared with the saline group (P<0.01; Fig. 4A and B). Of note, the increase in
spinal p-STAT3 expression by chronic morphine was markedly
inhibited by i.t. administration of spinal LA (3 μg), suggesting
that spinal STAT3 activation is modulated by the spinal leptin
following chronic morphine treatment. LA (3 μg) alone did not alter
the basal expression of spinal p-STAT3 (Fig. 4A and B). In addition, p-STAT3
immunoreactivity was topographically increased in the spinal dorsal
horn of chronic morphine treatment rats (Fig. 4C). The spinal p-STAT3
immunoreactivity was colocalized primarily with GFAP (a marker of
astrocytes; Fig. 4D).
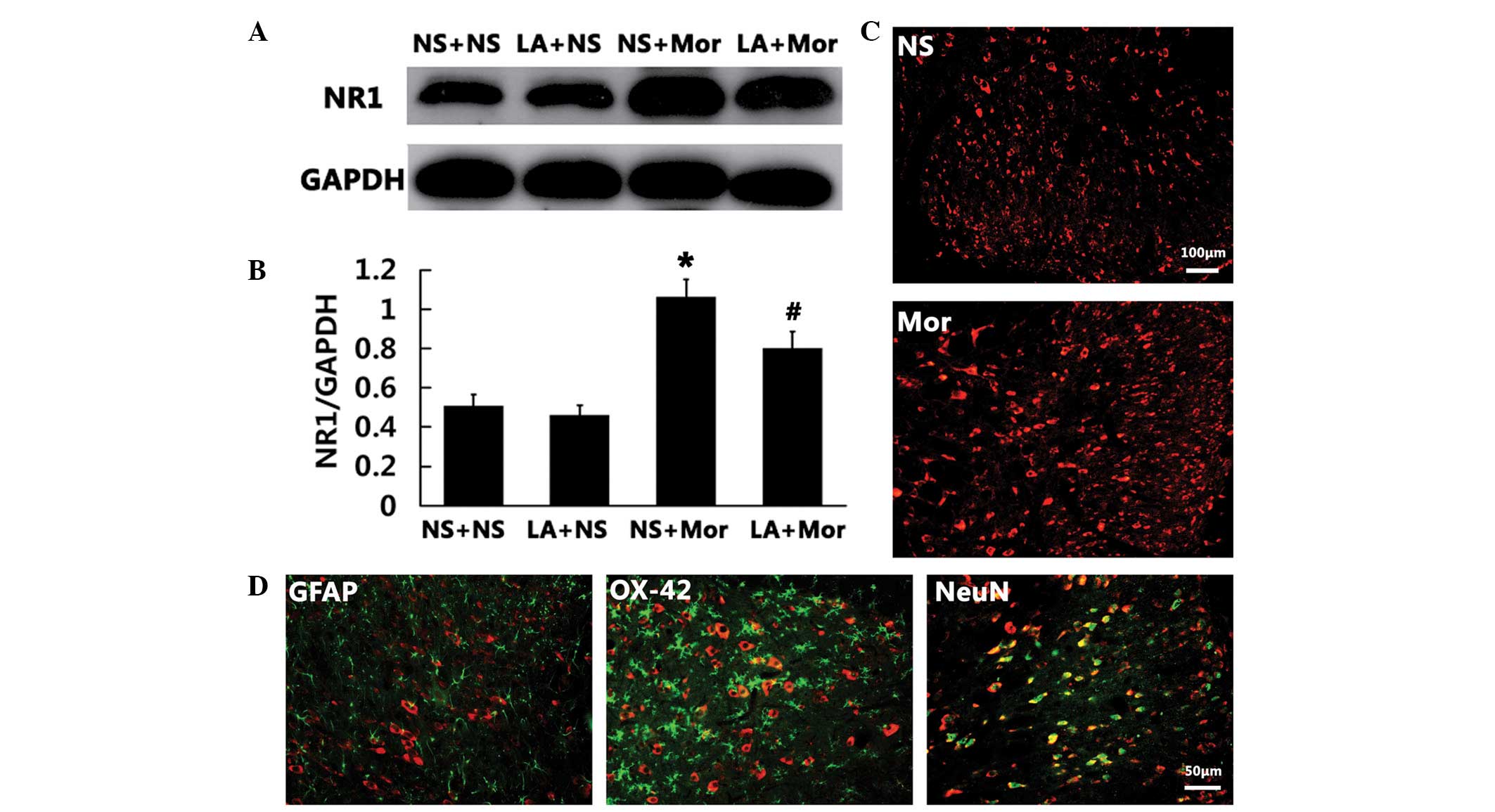
LA attenuates chronic morphine-induced
increase in the spinal NR1 subunit of NMDA receptors in rats
Following this, we examined whether there was a link
between the spinal NR1 subunit expression and the spinal leptin
following chronic morphine exposure. The findings of the western
blot analysis demonstrated that the expression of spinal NR1 was
markedly increased on day 7 following chronic morphine treatment as
compared with that in the saline group (P<0.01; Fig. 5A and B). Of note, the increased
spinal NR1 expression induced by chronic morphine treatment was
markedly inhibited by i.t. administration of LA (3 μg), suggesting
that the spinal NR1 expression is modulated by the spinal leptin
following chronic morphine treatment. LA (3 μg) alone did not alter
the basal expression of spinal NR1 (Fig. 5A and B). In addition, NR1
immunoreactivity was topographically increased in the spinal cord
dorsal horn in the chronic morphine treatment rats (Fig. 5C). Double immunofluorescence
results revealed that the spinal NR1 immunoreactivity was
colocalized primarily with NeuN (a neuronal marker; Fig. 5D).
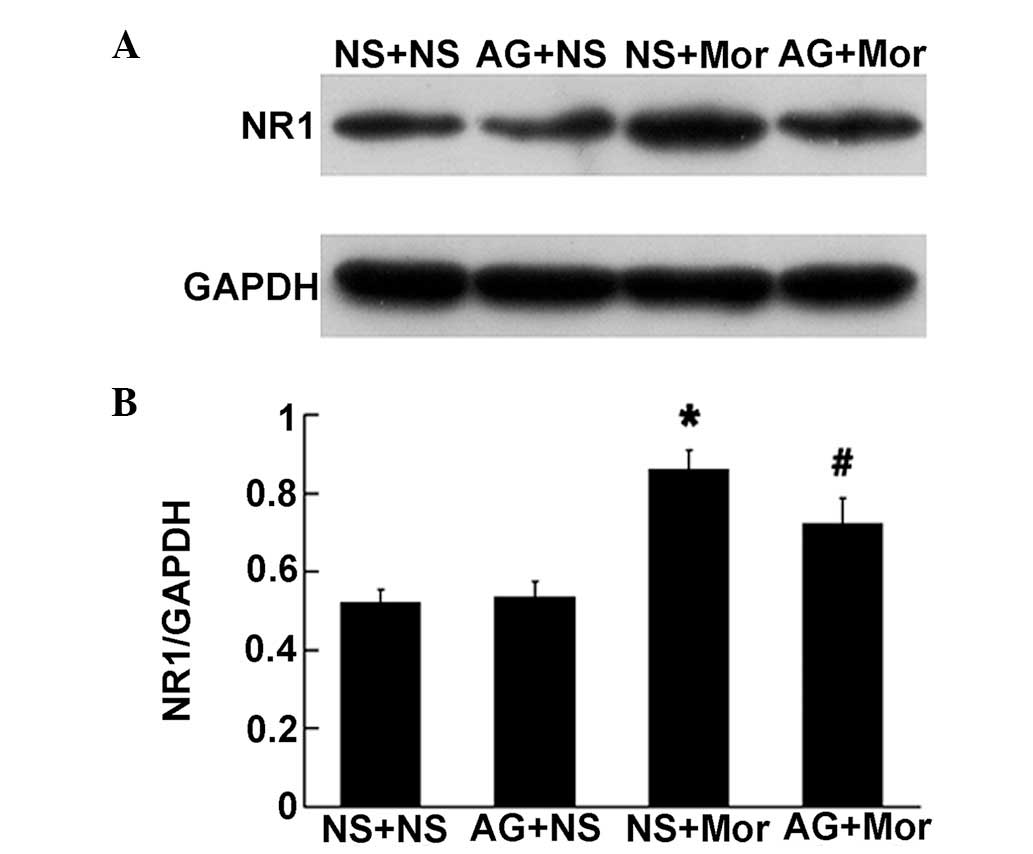
JAK-STAT inhibitor ameliorates chronic
morphine-induced upregulation of the spinal NR1 subunit of NMDA
receptors
Next, we observed whether the spinal STAT pathway
was implicated in chronic morphine-induced increase in spinal NR1
subunit expression. As illustrated in Fig. 6, pretreatment with 1 μg AG490 (an
inhibitor of JAK-STAT) for 30 min prior to chronic morphine
treatment, markedly attenuated the increased expression of spinal
NR1 subunit induced by chronic morphine treatment, suggesting the
spinal STAT pathway is involved in this upregulation process. AG490
(1 μg) alone failed to alter the basal expression of spinal NR1
(Fig. 6).
JAK-STAT inhibitor and NMDA receptor
inhibitor block the development of morphine antinociceptive
tolerance in rats
Finally, we determined the role of the JAK-STAT
pathway and NMDA receptors in the spinal leptin-mediated
development of morphine antinociceptive tolerance. As summarized in
Fig. 7, pretreatment with either 1
μg AG490 (an inhibitor of JAK-STAT) or 10 nM MK-801, (a
non-competitive antagonist of NMDA receptor) for 30 min prior to
chronic morphine treatment, markedly blocked the development of
morphine antinociceptive tolerance on days 3, 5 and 7, as compared
with the morphine treatment group, respectively. However, 1 μg
AG490 or 10 nM MK-801 alone did not induce any analgesic effect as
compared with the saline group (Fig.
7). Combined with the above results of this study (Figs. 1, 3–6),
these findings revealed that spinal leptin contributes to the
development of morphine antinociceptive tolerance in rats, at least
in part by activating the spinal STAT3-NMDA receptors pathway.
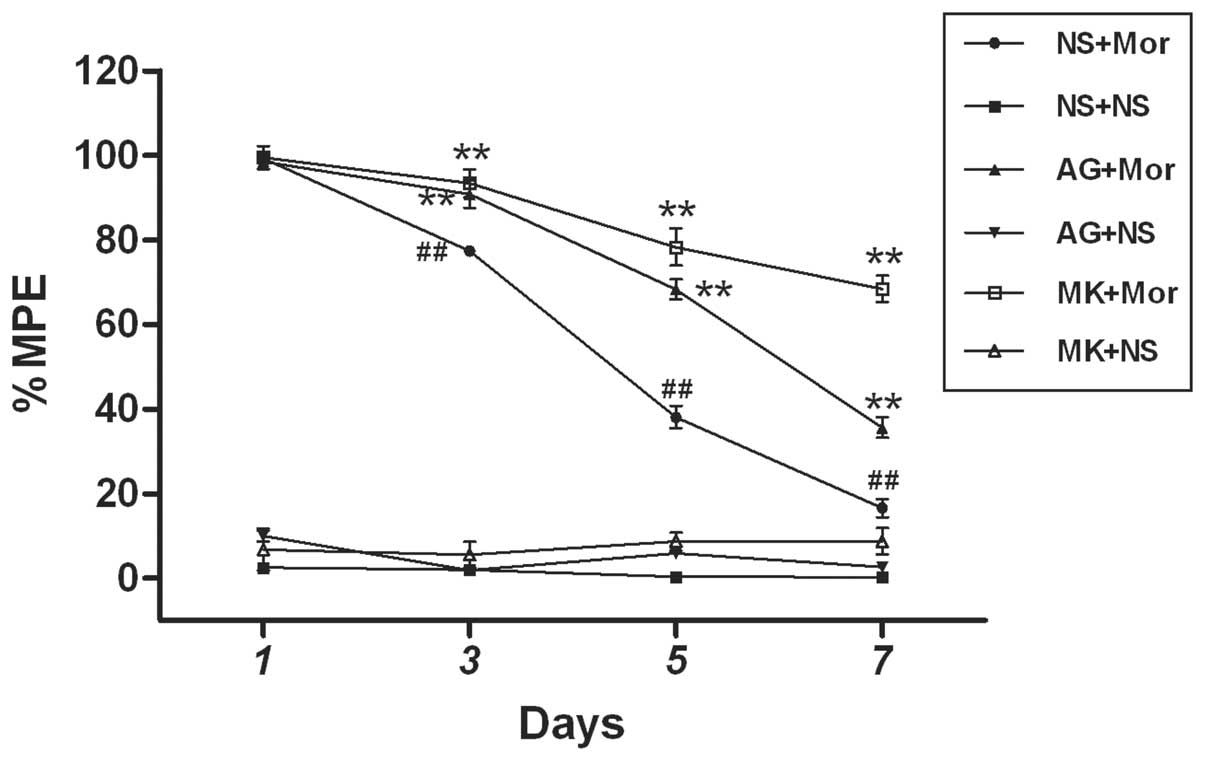 | Figure 7JAK-STAT inhibitor and NMDA receptor
antagonist attenuate the development of Mor antinociceptive
tolerance in rats. JAK-STAT inhibitor (AG490, 1 μg) or NMDA
receptor antagonist (MK-801, 10 nM) was administered 30 min prior
to daily Mor (15 μg) i.t. injection. Data are presented as the mean
± SEM (n=6). **P<0.001 vs. (NS+Mor) group,
##P<0.001 vs. (NS+ NS) group. NMDA,
N-methyl-D-aspartate; JAK-STAT, Janus kinase signal transducer and
activator of transcription; %MPE, the percentage of maximal
possible antinociceptive effect; NS, normal saline; Mor, morphine;
AG, AG490; MK, MK-801. |
Discussion
In the present study, novel data were provided,
demonstrating that spinal leptin contributes to the development of
morphine antinociceptive tolerance through the activation of the
STAT3-NMDA receptor pathway in rats. This is supported by the
following findings: (i) chronic morphine treatment upregulated the
expression levels of the spinal Ob-R and leptin in a time-dependent
manner; (ii) chronic morphine enhanced the spinal expression of
p-STAT3; (iii) chronic morphine increased the spinal expression
level of NR1 subunit of NMDA receptor; (iv) a LA blocked the
development of tolerance to morphine analgesia; (v) chronic
morphine-induced upregulation of the spinal expression of p-STAT3
and NR1 subunit of NMDA receptor was also attenuated by a LA; (vi)
chronic morphine-induced upregulation of NR1 subunit expression was
attenuated by AG490 (an inhibitor of JAK-STAT pathway) and (vii)
AG490 and MK-801 (a non-competitive antagonist of NMDA receptor)
reduced the development of morphine antinociceptive tolerance.
Leptin, the protein encoded by the obese (ob) gene,
was originally considered to be an adipose-derived hormone
(18). Recent studies, however,
have reported that leptin is produced by a variety of tissues,
including gastric mucosa, bone marrow, mammary epithelium, skeletal
muscle (19) and the heart
(20). In addition, leptin mRNA
expression has also been identified in a variety of brain regions
(21,22). In the present study, we observed
the expression of the spinal leptin protein and Ob-R expression, as
well as their immunoreactivity, at a basal level in the spinal cord
dorsal horn of rats. These data are consistent with previous
results that there is a basal expression of the leptin mRNA and
protein, as well as the long form of the leptin receptor (Ob-Rb),
within the spinal cord dorsal horn (11). Based on previous investigations
(11) and our studies, it was
hypothesized that the spinal cord dorsal horn of rat is a site of
leptin production and action.
Of note, leptin has been demonstrated to contribute
to the pathogenesis of neuropathic pain (10–12).
However, whether leptin is involved in the development of morphine
antinociceptive tolerance is unclear. Since it is hypothesized that
morphine tolerance and neuropathic pain share some common
pathological mechanisms (13,14),
this prompted us to examine the role of spinal leptin in the
development of morphine antinociceptive tolerance. The results
demonstrated, for the first time, to the best of our knowledge,
that the spinal leptin critically contributes to the development of
morphine antinociceptive tolerance because (i) i.t. administration
of a LA significantly attenuated the development of morphine
antinociceptive tolerance; (ii) the expression of spinal leptin
protein and immunoreactivity was markedly enhanced after chronic
morphine treatment and (iii) the expression of spinal Ob-R protein
and immunoreactivity was evidently upregulated following chronic
morphine treatment. However, the underlying mechanisms of the
spinal leptin upregulation following chronic morphine remain
elusive and further studies are required to investigate this.
To examine the molecular mechanisms underlying the
role of spinal leptin in the development of morphine
antinociceptive tolerance, we investigated whether the activation
of STAT3 mediates spinal leptin-induced morphine antinociceptive
tolerance. This was because activation of the JAK-STAT pathway has
been demonstrated, via targeted gene transcriptions, to be
responsible for a number of leptin functions, that were indicative
of a genomic mechanism of action (23,24).
Leptin binds to the extracellular domain of the Ob-Rb dimmer,
activating the JAK2 tyrosine kinase. The activated JAK2 tyrosine
kinase phosphorylates itself, Tyr1138 and Tyr1007 on the
intracellular tail of Ob-Rb (17).
The p-Tyr1138 binds and mediates the phosphorylation-dependent
activation of STAT3. STAT3 then translocates to the nucleus to
activate the transcription of downstream target genes. Recently,
Lim et al reported that spinal leptin mediates neuropathic
pain induced by chronic constriction sciatic nerve injury (CCI) by
activating the JAK-STAT pathway, and that the i.t. leptin
induced-pain behavioral and cellular changes (such as the
upregulation of NR1 subunit of NMDA receptors and IL-1β expression)
are markedly ameliorated by coadministration of AG490 (a JAK/STAT
inhibitor) (11). In this study,
consistent with a recent study reported by Wang et al
(5), we demonstrated that the
expression of spinal p-STAT protein (western blot analysis) and
immunoreactivity, were significantly upregulated following chronic
morphine treatment. The increased spinal p-STAT3 expression was
attenuated by i.t. injection of a LA. In addition, pretreatment
with AG490, prior to exposure to chronic morphine, markedly
depressed the development of morphine antinociceptive tolerance.
These results suggest that the activation of STAT3 is implicated in
spinal leptin-mediated development of morphine antinociceptive
tolerance, which extends the findings of a recent study (5).
NMDA receptors have been demonstrated to contribute
to the pathogenesis of neuropathic pain (11,12,25,26)
and tolerance to morphine analgesia (1,2,27).
Furthermore, there appears to be a functional link between leptin
and NMDA receptors. For example, exposure to leptin enhances NMDA
receptor activity and modulates NMDA receptor-dependent long term
potential (LTP) in an in vitro hippocampal preparation
(28). I.t. administration of
MK-801 attenuates exogenous leptin-induced thermal hyperalgesia and
mechanical allodynia (12).
More recently, a study also demonstrated that leptin
upregulates the expression of spinal NMDA receptor via the JAK/STAT
pathway (11). Based on these
previous studies (1,2,11,12,25–28),
we investigated the role of the NR1 subunit of the NMDA receptor in
spinal leptin-mediated development of morphine antinociceptive
tolerance. Our findings demonstrated that the activation of
STAT3-NMDA receptor pathway is an important component of this
process in rats. This is supported by the following results that
(i) the level of spinal NR1 subunit protein and immunoreactivity
was enhanced following chronic morphine administration, (ii) the
increased expression of spinal NR1 was attenuated by a LA, (iii)
the increased expression of spinal NR1 was diminished by AG490 (a
JAK/STAT inhibitor) and (iv) MK-801, a non-competitive antagonist
of NMDA receptor considerably blocked the development of morphine
antinociceptive tolerance. However, a regulatory effect of
descending neural pathways may not be excluded in our in
vivo experiments. Therefore more studies, including in
vitro experiments, are required to more accurately define
whether the modulatory effects of spinal leptin on the activation
of spinal STAT3-NR1 pathway were associated with its direct or
indirect spinal action, or a combination of both.
To date, multiple factors have been demonstrated to
contribute to the development of tolerance to morphine analgesia.
Our results reveal that the spinal leptin-STAT3-NMDA pathway may be
a unique target for new pharmacological interventions for patients
exhibiting morphine tolerance, because leptin has been demonstrated
to have a critical role in a number of neuronal functions,
including neuroplasticity and neuroendocrine regulation (29). Further studies examining the role
of other signaling cascades used by leptin, may facilitate our
understanding of the mechanisms responsible for the contribution of
leptin to the development of morphine antinociceptive
tolerance.
Acknowledgements
This study was supported by grants from the Science
and Technology Planning Project of Guangdong of China
(2012B031800358) and the National Natural Science Foundation of
China (no. 30800335 and no. 81100827).
References
|
1
|
Trujillo KA and Akil H: Inhibition of
morphine tolerance and dependence by the NMDA receptor antagonist
MK-801. Science. 251:85–87. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Guo RX, Zhang M, Liu W, Zhao CM, Cui Y,
Wang CH, Feng JQ and Chen PX: NMDA receptors are involved in
upstream of the spinal JNK activation in morphine antinociceptive
tolerance. Neurosci Lett. 467:95–99. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Raghavendra V, Rutkowski MD and DeLeo JA:
The role of spinal neuroimmune activation in morphine
tolerance/hyperalgesia in neuropathic and sham-operated rats. J
Neurosci. 22:9980–9989. 2002.PubMed/NCBI
|
|
4
|
Johnston IN, Milligan ED, Wieseler-Frank
J, Frank MG, Zapata V, Campisi J, Langer S, Martin D, Green P,
Fleshner M, et al: A role for proinflammatory cytokines and
fractalkine in analgesia, tolerance, and subsequent pain
facilitation induced by chronic i.t morphine. J Neurosci.
24:7353–7365. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Z, Ma W, Chabot JG and Quirion R:
Calcitonin gene-related peptide as a regulator of neuronal
CaMKII-CREB, microglial p38-NFκB and astroglial ERK-Stat1/3
cascades mediating the development of tolerance to morphine-induced
analgesia. Pain. 151:194–205. 2010.PubMed/NCBI
|
|
6
|
Cui Y, Liao XX, Liu W, Guo RX, Wu ZZ, Zhao
CM, Chen PX and Feng JQ: A novel role of minocycline: attenuating
morphine antinociceptive tolerance by inhibition of p38 MAPK in the
activated spinal microglia. Brain Behav Immun. 22:114–123. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ and
Chen PX: Activation of p38 mitogen-activated protein kinase in
spinal microglia mediates morphine antinociceptive tolerance. Brain
Res. 1069:235–243. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Proenca R, Maffei M, Barone M,
Leopold L and Friedman JM: Positional cloning of the mouse obese
gene and its human homologue. Nature. 372:425–432. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bjørbaek C, Uotani S, da Silva B and Flier
JS: Divergent signaling capacities of the long and short isoforms
of the leptin receptor. J Biol Chem. 272:32686–32695.
1997.PubMed/NCBI
|
|
10
|
Maeda T, Kiguchi N, Kobayashi Y, Ikuta T,
Ozaki M and Kishioka S: Leptin derived from adipocytes in injured
peripheral nerves facilitates development of neuropathic pain via
macrophage stimulation. Proc Natl Acad Sci USA. 106:13076–13081.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lim G, Wang S, Zhang Y, Tian Y and Mao J:
Spinal leptin contributes to the pathogenesis of neuropathic pain
in rodents. J Clin Invest. 119:295–304. 2009.PubMed/NCBI
|
|
12
|
Tian Y, Wang S, Ma Y, Lim G, Kim H and Mao
J: Leptin enhances NMDA-induced spinal excitation in rats: A
functional link between adipocytokine and neuropathic pain. Pain.
152:1263–1271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mao J, Price DD and Mayer DJ: Mechanisms
of hyperalgesia and morphine tolerance: a current view of their
possible interactions. Pain. 62:259–274. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mayer DJ, Mao J, Holt J and Price DD:
Cellular mechanisms of neuropathic pain, morphine tolerance, and
their interactions. Proc Natl Acad Sci USA. 96:7731–7736. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harvey J: Leptin: a diverse regulator of
neuronal function. J Neurochem. 100:307–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tartaglia LA, Dembski M, Weng X, et al:
Identification and expression cloning of a leptin receptor, OB-R.
Cell. 83:1263–1271. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Myers MG Jr: Leptin receptor signaling and
the regulation of mammalian physiology. Recent Prog Horm Res.
59:287–304. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ahima RS, Qi Y, Singhal NS, Jackson MB and
Scherer PE: Brain adipocytokine action and metabolic regulation.
Diabetes. 55(Suppl 2): S145–154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wolsk E, Mygind H, Grøndahl TS, Pedersen
BK and van Hall G: Human skeletal muscle releases leptin in vivo.
Cytokine. 60:667–673. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Purdham DM, Zou MX, Rajapurohitam V and
Karmazyn M: Rat heart is a site of leptin production and action. Am
J Physiol Heart Circ Physiol. 287:H2877–H2884. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Morash B, Li A, Murphy PR, Wilkinson M and
Ur E: Leptin gene expression in the brain and pituitary gland.
Endocrinology. 140:5995–5998. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ur E, Wilkinson DA, Morash BA and
Wilkinson M: Leptin immunoreactivity is localized to neurons in rat
brain. Neuroendocrinology. 75:264–272. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bjorbaek C: Central leptin receptor action
and resistance in obesity. J Investig Med. 57:789–794.
2009.PubMed/NCBI
|
|
24
|
Robertson SA, Leinninger GM and Myers MG
Jr: Molecular and neural mediators of leptin action. Physiol Behav.
94:637–642. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fisher K, Coderre TJ and Hagen NA:
Targeting the N-methyl-D-aspartate receptor for chronic pain
management. Preclinical animal studies, recent clinical experience
and future research directions. J Pain Symptom Manage. 20:358–373.
2000. View Article : Google Scholar
|
|
26
|
Hewitt DJ: The use of NMDA-receptor
antagonist in the treatment of chronic pain. Clin J Pain.
16(Suppl): s73–79. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Houghton AK, Parsons CG and Headley PM:
Mrz 2/579, a fast kinetic NMDA channel blocker, reduces the
development of morphine tolerance in awake rats. Pain. 91:201–207.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goldin M, Segal M and Avignone E:
Functional plasticity triggers formation and pruning of dendritic
spines in cultured hippocampal networks. J Neurosci. 21:186–193.
2001.PubMed/NCBI
|
|
29
|
Elmquist JK, Maratos-Flier E, Saper CB and
Flier JS: Unraveling the central nervous system pathways underlying
responses to leptin. Nat Neurosci. 1:445–450. 1998. View Article : Google Scholar : PubMed/NCBI
|