Introduction
Glycyrrhiza Radix has been used as a treatment for
thousands of years in China and its major components have been
reported to exhibit various pharmacological activities, including
anti-inflammatory (1), -obesity
(2), -viral (3), -oxidative (4) and neuroprotective (5) effects. Liquiritin (LQ), one of the
major compounds extracted from Glycyrrhiza Radix, possesses
anti-depressant-like effects, as has been indicated by
tail-suspension and forced swimming tests in mice (6). LQ also exerts neurotrophic effects,
whereby it promotes nerve growth factor (NGF)-induced neurite
outgrowth (7). The chemical
structure of LQ is shown in Fig.
1. A previous study has reported that LQ may exert
neuroprotective effects in cerebral ischemia/reperfusion-induced
brain damage through antioxidant and anti-apoptotic mechanisms
(8). However, the neuroprotective
effect of LQ against glutamate-induced cell damage has not yet been
elucidated.
Glutamate, an important neurotransmitter in the
vertebrate nervous system, has a key role in learning and memory
(9). Glutamate-mediated
excitotoxicity occurs as part of the ischemic cascade (10) and is associated with numerous
diseases, including amyotrophic lateral sclerosis, autism,
Alzheimer’s disease and certain forms of mental retardation
(9). Several signaling pathways
are involved in the regulation of glutamate-induced neurotoxicity
(11,12). Extracellular signal-regulated
kinases (ERKs) and AKT signaling pathways have been proposed to
contribute to cell differentiation, proliferation, survival and
apoptosis (13–15). Furthermore, previous studies have
demonstrated that glutamate significantly downregulates AKT and ERK
phosphorylation (16,17). A previous study has also shown that
sodium ferulate protects cortical neurons against glutamate-induced
apoptosis through phosphatidylinositide 3-kinase (PI3K)/AKT and ERK
signaling pathways (17).
In the present study, LQ was found to protect
differentiated PC12 (DPC12) cells against glutamate-induced reduced
cell viability, high apoptosis rates, excessive lactate
dehydrogenase (LDH) release, intracellular Ca2+ overload
and mitochondrial dysfunction. Furthermore, LQ pretreatment was
observed to normalize the glutamate-induced alterations in pro- and
anti-apoptotic protein expression. The LQ-mediated neuroprotective
effect against glutamate-induced DPC12 cell damage was found to be
associated with ERK and AKT activation.
Materials and methods
Cell lines and culture
PC12 cells (CRL-1721; American Type Culture
Collection, Rockville, MD, USA) were used at passages <10 and
were maintained as monolayer cultures in Dulbecco’s Modified Eagle
Medium (DMEM) supplemented with 10% horse serum (HS; Invitrogen
Life Technologies, Carlsbad, CA, USA), 5% fetal bovine serum (FBS;
Invitrogen Life Technologies), 100 U/ml penicillin and 100 μg/ml
streptomycin in a humidified atmosphere containing 5%
CO2 and 95% air at 37°C. Cells were differentiated using
the addition of 20 ng/ml NGF (Sigma-Aldrich, St. Louis, MO, USA) in
DMEM supplemented with 1% HS, 1% FBS, 100 U/ml penicillin and 100
μg/ml streptomycin for 48 h.
Cell viability assay
Cell viability was measured using a quantitative
colorimetric assay with MTT (Sigma-Aldrich) as described previously
(18). Briefly, PC12 cells were
seeded onto 96-well plates at a density of 2×104/well
and differentiated using NGF. Cells were pretreated with 25 and 50
μM LQ (purity >98.0%; Shanghai Source Leaves Biological
Technology Co., Ltd., Shanghai, China) for 3 h and co-treated with
20 mM glutamate for 24 h. In separate experiments, DPC12 cells
underwent 30 min pretreatment with 10 μM PD98059, an ERK inhibitor,
or 10 μM LY294002, a PI3K inhibitor. Cells were then treated with
25 or 50 μM LQ for 3 h, prior to exposure to 20 mM glutamate for 24
h. Treated cells were subsequently incubated with MTT solution (0.5
mg/ml) for 4 h at 37°C in the dark. The absorbance was measured
using a microplate reader (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) at 540 nm. The viability of the treated cells was
expressed as a percentage of that of the corresponding control
cells.
Released LDH analysis
The In Vitro Toxicology Assay kit
(Sigma-Aldrich) was used to detect LDH release in the culture
medium. PC12 cells were seeded onto six-well plates at a density of
1×105/well and were differentiated using NGF. DPC12
cells were pretreated with 25 and 50 μM LQ for 3 h and then
co-treated with 20 mM glutamate for 24 h. The medium in each
treatment group was collected individually. A total of 60 μl mixed
assay solution was added to 30 μl culture medium. Following
incubation at room temperature in the dark for 30 min, 10 μl 1 N
HCl was added to terminate the reaction. Absorbance was
spectrophotometrically measured at a wavelength of 490 nm. LDH
release in the treatment groups was expressed as a percentage of
the LDH released in the control group.
Flow cytometric analysis of
apoptosis
Annexin V and propidium iodide (PI) double staining
was used to determine alterations in cell apoptosis. PC12 cells
were seeded onto six-well plates at a density of
1×105/well and differentiated. DPC12 cells were then
pretreated with 25 and 50 μM LQ for 3 h, prior to co-treatment with
20 mM glutamate for 24 h. Subsequent to collection, cells were
suspended in binding buffer containing 20 μg/ml Annexin
V-fluorescein isothiocyanate and 50 μg/ml PI, and incubated for 20
min at room temperature. Cell apoptosis rate was analyzed using a
flow cytometer (FC500; Beckman Coulter, Inc., Brea, CA, USA).
Intracellular Ca2+
concentration analysis
Cells were stained with Fluo-4 AM (Invitrogen Life
Technologies) at a final concentration of 5 μM in order to
determine the intracellular Ca2+ concentration. PC12
cells were seeded onto confocal dishes at a density of
1×105 cells/well and differentiated. Subsequent to
pretreatment with 25 μM LQ for 3 h and co-treatment with 20 mM
glutamate for 12 h, cells were incubated with Fluo-4 AM for 30 min
at 37°C in the dark. Following three washes with phosphate-buffered
saline (PBS), the fluorescence intensity was determined using laser
scanning confocal microscopy (Axio Observer Z1; Carl Zeiss,
Oberkochen, Germany) with an excitation wavelength of 488 nm and an
emission wavelength of 520 nm at a magnification of ×20.
Mitochondrial membrane potential (Δψm)
analysis
5,5′,6,6′-Tetrachloro-1,1′,3,3′
tetraethylbenzimidazolylcarbocyanine iodide (JC-1; Sigma-Aldrich)
staining was used to examine alterations in Δψm. PC12 cells were
seeded onto confocal dishes at a density of 1×105
cells/well and differentiated. Subsequent to pretreatment with 25
μM LQ for 3 h and co-treatment with 20 mM glutamate for 12 h, cells
were incubated with 2 μM JC-1 at 37°C for 10 min in the dark.
Following three washes with PBS, changes in mitochondrial
fluorescence were examined using a fluorescent microscope (Axio
Observer Z1; Carl Zeiss) at a magnification of ×20. Red
fluorescence was observed in healthy cells with a high Δψm and
green fluorescence was apparent in apoptotic or unhealthy cells
with a low Δψm (19).
Western blot analysis
Treated cells were lysed in radioimmunoprecipitation
assay buffer containing 1% protease inhibitor cocktail and 2%
phenylmethanesulfonyl fluoride (Sigma-Aldrich). In order to detect
cytochrome c (cyto c) release, cytoplasmic extracts
were prepared as described previously by Yang et al
(20). A total of 30 μg protein
was separated using 10–12% SDS-PAGE and electrophoretically
transferred onto nitrocellulose membranes (pore size, 0.45 μm; Bio
Basic, Inc., Markham, ON, Canada). The transferred membranes were
then blotted with antibodies against phosphorylated (P)-ERKs, total
(T)-ERKs, P-AKT, T-AKT, P-glycogen synthase kinase-3β (GSK3β),
T-GSK3β, B-cell lymphoma 2 (Bcl-2), Bcl2-associated X protein
(Bax), cyto c and GAPDH at dilutions of 1:1,000 (Cell
Signaling Technology, Inc., Danvers, MA, USA) at 4°C overnight.
Membranes were then incubated with horseradish
peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) for 3 h at 4°C.
Chemiluminescence was detected using enhanced chemiluminescence
detection kits (GE Healthcare, Amersham, UK). The intensity of the
bands was quantified by scanning densitometry using Quantity One
4.5.0 software (Bio-Rad Laboratories, Inc.).
Statistical analysis
One-way analysis of variance was used to detect
statistical significance, followed by post hoc multiple comparison
tests. Data are expressed as the mean ± standard deviation. A value
of P<0.05 was considered to indicate a statistically significant
difference.
Results
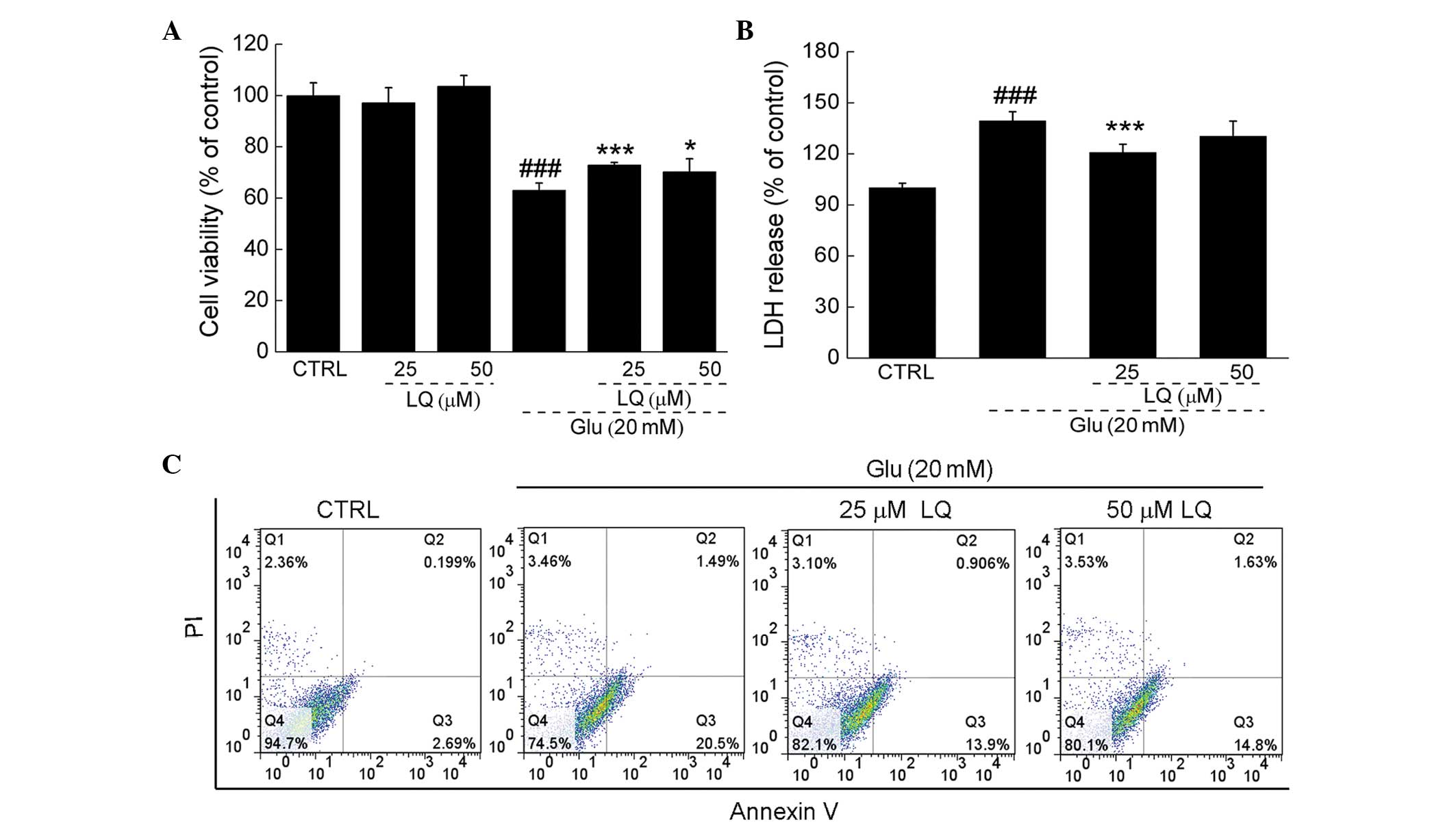
LQ protects DPC12 cells from
glutamate-induced apoptotic cell damage
Exposure of DPC12 cells to 20 mM glutamate for 24 h
resulted in ~38% cell death; however, upon pretreatment with 25 or
50 μM LQ for 3 h, cell death was significantly reduced (71 and 74%
viability vs. 62% viability, P<0.05). Pretreatment with 25 and
50 μM LQ alone showed no effect on cell proliferation (Fig. 2A).
In DPC12 cells exposed to 20 mM glutamate, LDH
release was observed to be 39% greater than that in the control
cells (P<0.001). However, pretreatment with 25 μM LQ was found
to significantly suppress LDH release to levels 20% higher than
those in the control cells (139 vs. 120%, P<0.001) (Fig. 2B). Furthermore, flow cytometry
revealed that LQ reduced the proportion of apoptotic cells compared
with the cells solely exposed to glutamate (Fig. 2C).
LQ attenuates intracellular
Ca2+ overload and restores the dissipation of Δψm
Fluo-4 AM staining was used to assess the changes in
Ca2+ concentration in DPC12 cells. In cells exposed to
20 mM glutamate for 12 h, high Ca2+ influx was observed,
as indicated by the increase in fluorescence intensity.
Pretreatment with 25 μM LQ was found to reduce this Ca2+
overload (Fig. 3A).
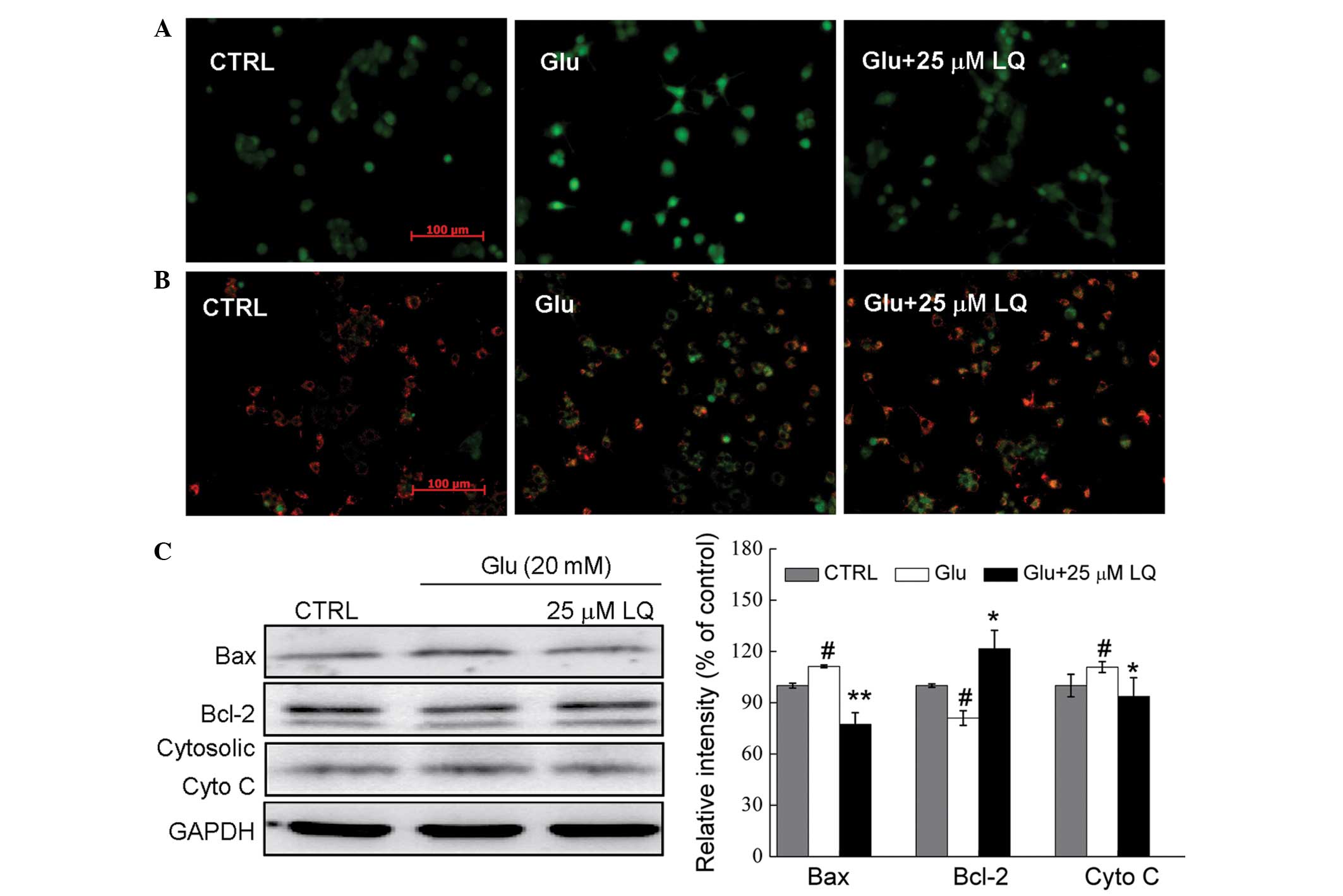 | Figure 3In Glu-exposed DPC12 cells, LQ
restores (A) intracellular Ca2+ overload (magnification,
×20), (B) mitochondrial membrane potential dissipation
(magnification, ×20) and (C) alterations in the expression of
apoptosis-related proteins. Cells were pretreated with 25 μM LQ for
3 h and exposed to 20 mM Glu for (A and B) 12 h or (C) 24 h. Bcl-2,
Bax and cytosolic cyto c expression was normalized using
GAPDH. Data are expressed as a percentage of the value in the
corresponding control group and are presented as the mean ±
standard deviation of three replicate experiments.
#P<0.05 vs. control group; *P<0.05 and
**P<0.01 vs. Glu-treated cells. LQ, liquiritin; Glu,
glutamate; Bcl-2, B-cell lymphoma-2; Bax, Bcl2-associated X
protein; cyto c; cytochrome c; CTRL, control. |
Mitochondrial function is one of the factors
responsible for cell apoptosis. JC-1 staining revealed that
pretreatment with 25 μM LQ (21)
significantly restored the glutamate-induced dissipation of Δψm, as
indicated by an increase in red fluorescence in the LQ-pretreated
cells compared with those treated solely with glutamate (Fig. 3B).
Glutamate exposure was found to enhance Bax
expression by 11%, reduce Bcl-2 expression by 20% and increase
cytosolic cyto c expression by 10% compared with the
non-treated control cells (all P<0.05). However, LQ markedly
reduced the glutamate-induced increase in Bax and cytosolic cyto
c expression to normal levels, and enhanced the expression
of Bcl-2 (P<0.05) (Fig.
3C).
ERK and AKT/GSK3β activation contributes
to LQ-mediated neuroprotection in DPC12 cells
ERK and AKT/GSK3β activation was detected in DPC12
cells. While glutamate exposure for between 30 and 360 min was
found to significantly inhibit ERK phosphorylation, exposure to 25
μM LQ alone for 60 and 180 min was found to significantly enhance
the expression of P-ERKs (P<0.05). Furthermore, pretreatment
with LQ for between 60 and 360 min was observed to significantly
reverse the glutamate-induced suppression of P-ERK expression
(P<0.05) (Fig. 4A and B).
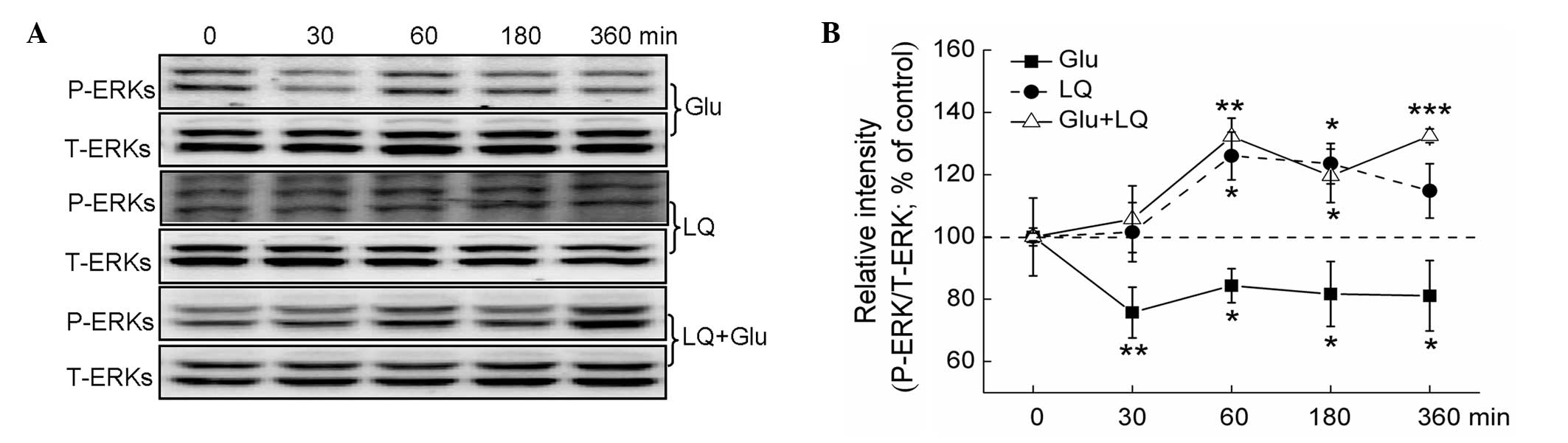 | Figure 4ERK pathways are involved in
LQ-mediated neuroprotection against Glu-induced cell damage. DPC12
cells were treated with LQ or Glu alone and collected at 0, 30, 60,
180 and 360 min. For LQ and Glu co-treatment, DPC12 cells were
pretreated with 25 μM LQ for 3 h, followed by Glu. Cells were then
collected at 0, 30, 60, 180 and 360 min subsequent to Glu exposure
(A) Expression of P-ERKs and T-ERKs detected using western blot
analysis. (B) Quantification of the expression of P-ERKs and
T-ERKs. The expression of P-ERKs was normalized using that of
T-ERKs. Data are presented as the mean ± standard deviation of
three replicate experiments. *P<0.05,
**P<0.01 and ***P<0.001 vs. cells
collected at 0 min. ERK, extracellular signal-regulated kinase;
Glu, glutamate; LQ, liquiritin; P-, phosphorylated; T-, total. |
PI3K/AKT are crucial regulators of
glutamate-mediated cell damage (17). Glutamate treatment for between 30
and 360 min was found to significantly suppress P-AKT and P-GSK3β
expression. Exposure to LQ alone and in combination with glutamate
resulted in a time-dependent increase in P-AKT and P-GSK3β
expression (P<0.05), but did not affect expression of T-AKT and
T-GSK3β (Fig. 5A–D).
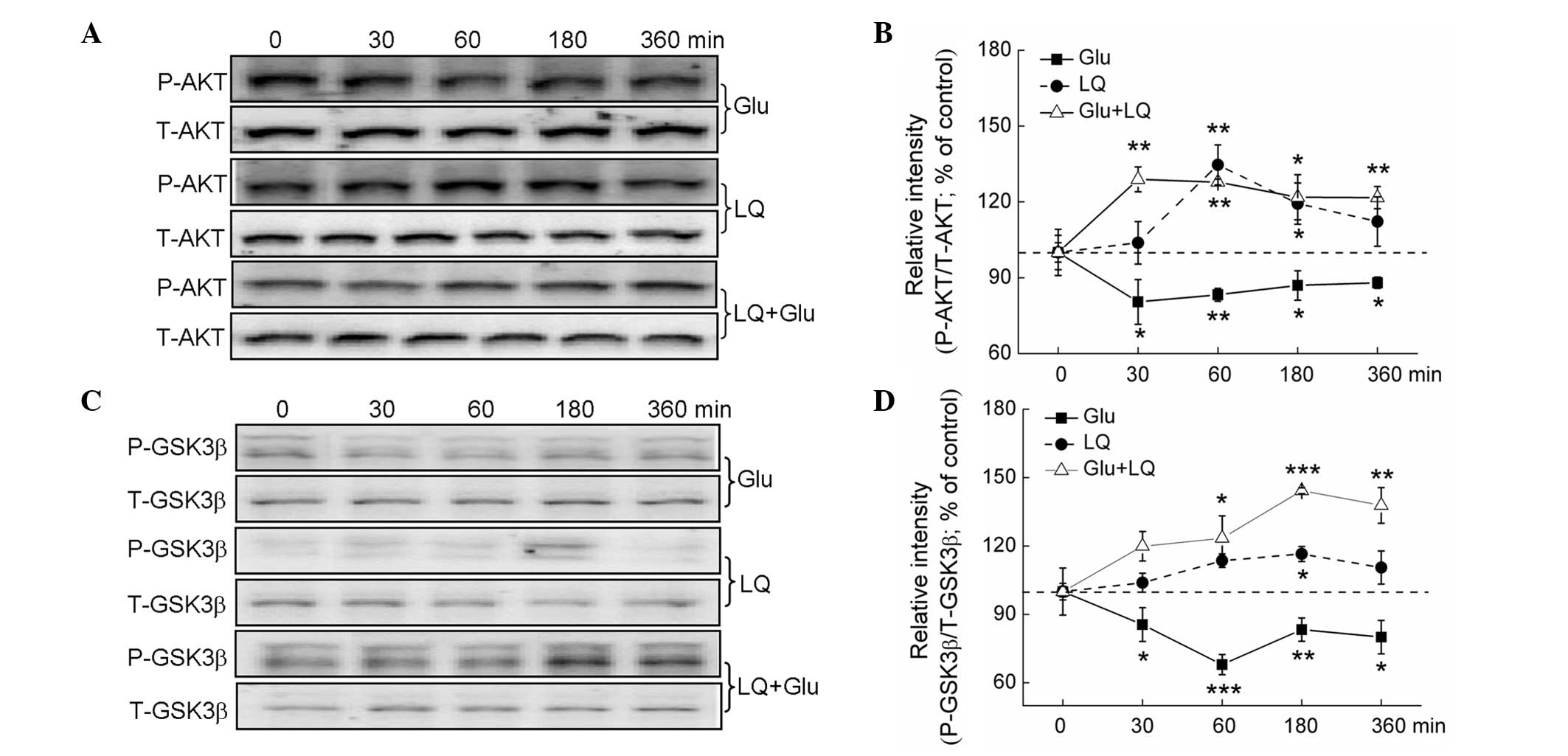 | Figure 5The AKT/GSK3β pathway contributes to
LQ-mediated neuroprotection against Glu-induced cell damage. DPC12
cells were treated with LQ or Glu alone and collected at 0, 30, 60,
180 and 360 min. For LQ and Glu co-treatment, DPC12 cells were
pretreated with 25 μM LQ for 3 h, followed by Glu. Cells were then
collected at 0, 30, 60, 180 and 360 min subsequent to Glu exposure.
(A and C) Expression of P-AKT, T-AKT, P-GSK3β and T-GSK3β detected
using western blot analysis. (B and D) Quantification of P-AKT and
P-GSK3β expression, normalized using T-AKT and T-GSK-3β expression,
respectively. Data are expressed as a percentage of the value in
the corresponding control group and presented as the mean ±
standard deviation of three replicate experiments.
*P<0.05, **P<0.01 and
***P<0.001 vs. cells collected at 0 min. GSK3β,
glycogen synthase kinase-3β; P-, phosphorylated; T-, total; LQ,
liquiritin; Glu, glutamate. |
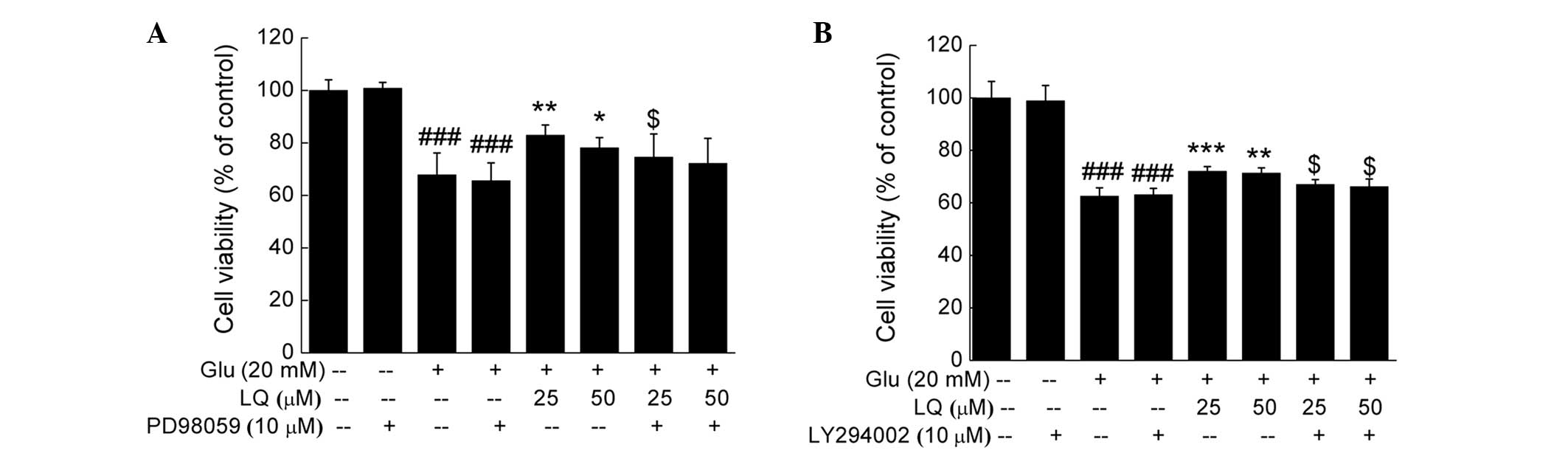
DPC12 cells underwent 30 min pretreatment with 10 μM
ERK or PI3K inhibitor, PD98059 or LY294002 respectively, followed
by 3 h treatment with LQ and 24 h exposure to glutamate. Treatment
with PD98059 or LY294002 did not affect cell viability compared
with the untreated or glutamate-treated cells; however, it was
found to significantly reduce the potency of LQ in enhancing cell
viability (P<0.05) (Fig.
6).
Discussion
The present study investigated the neuroprotective
effect of LQ against glutamate-induced cell damage and its
underlying mechanism. LQ was found to significantly attenuate the
glutamate-induced decrease in DPC12 cell viability and apoptotic
alterations, including mitochondrial function, the expression of
apoptosis-related proteins, intracellular Ca2+
concentration and LDH release. Furthermore, the activation of ERKs
and AKT/GSK-3β was found to contribute to LQ-mediated
neuroprotection.
Dissipation of Δψm and elevated mitochondrial cyto
c release were observed in glutamate-exposed DPC12 cells.
Experimental evidence has indicated that mitochondria have a key
role in executing important intracellular events associated with
neuronal survival and apoptosis (21). Certain apoptosis-related proteins,
including Bcl-2 and Bax, target the mitochondria and induce
mitochondrial swelling or increase the permeability of the
mitochondrial membrane. This leads to the efflux of apoptotic
effectors from the mitochondria (22,23).
Cyto c, released from mitochondria, serves as a regulatory
factor in morphological apoptosis-related changes (24). In the present study, after 3 h
pretreatment with LQ, the glutamate-induced dissipation of Δψm was
markedly restored and the expression of Bcl-2, Bax and cytosolic
cyto c was normalized. These findings indicate that the
neuroprotective effect of LQ may, at least partly, be attributed to
its restoration of Δψm through upregulation of the activity of
mitochondria-dependent apoptotic molecules.
AKT activation is associated with cell survival and
proliferation (25). GSK-3β, a
constitutively active enzyme substrate of AKT, is inactivated by
P-AKT (26). It has been reported
that GSK-3β inactivation is involved in the guanosine-mediated
protective effects against glutamate-induced cell death in SH-SY5Y
cells (26). Furthermore, GSK-3β
inhibition has been found to protect against ischemia/reperfusion
organ injury (27). In the present
study, exposure to LQ alone or in combination with glutamate was
observed to markedly enhance P-AKT and P-GSK3β levels in a
time-dependent manner in DPC12 cells compared with untreated cells.
In addition, pretreatment with the PI3K/AKT inhibitor LY294002 was
found to partially antagonize the LQ-induced increase in cell
viability. Furthermore, the increase in AKT activation observed
upon pretreatment with LQ resulted in an increase in GSK3β
phosphorylation, which has an important role in LQ-mediated
neuroprotection. Previous studies have suggested that the
activation of AKT regulates the expression of Bcl-2 (28). The AKT/Bcl-2 pathway contributes to
the protective effect of sodium ferulate in cultured cortical
neurons (17). Bcl-2 acts as an
upstream checkpoint of mitochondrial function (29); therefore, the findings of the
present study may indicate that mitochondrial function is
associated with AKT activation in LQ-exposed DPC12 cells.
ERKs were also analyzed in the present study.
Treatment with LQ alone or in combination with glutamate was found
to induce rapid phosphorylation of ERKs, whereas glutamate
treatment alone was observed to reduce P-ERK expression. PD98059
diminished the protective effect of LQ against the
glutamate-induced neurotoxicity and reduction in cell viability. It
has previously been reported that the inhibition of ERKs using a
specific inhibitor results in downregulation of Bcl-2 (30). These findings suggest that the
protective effect mediated by LQ may be achieved through ERK
pathways, which may be associated with mitochondrial function.
In conclusion, to the best of our knowledge, the
present study provides the first experimental evidence that LQ has
a neuroprotective effect against glutamate-induced cell damage, and
that this effect is associated with ERK and AKT/GSK3β pathways in
DPC12 cells. These findings suggest that LQ may have potential as a
therapeutic agent for the treatment of neurodegenerative diseases
and neural injury.
Acknowledgements
This study was supported by a grant from the
National Science and Technology support program of P.R. China
(grant no. 2012BAL29B05).
References
|
1
|
Wang CY, Kao TC, Lo WH and Yen GC:
Glycyrrhizic acid and 18β-glycyrrhetinic acid modulate
lipopolysaccharide-induced inflammatory response by suppression of
NF-κB through PI3K p110δ and p110γ inhibitions. J Agric Food Chem.
59:7726–7733. 2011.
|
|
2
|
Birari RB, Gupta S, Mohan CG and Bhutani
KK: Antiobesity and lipid lowering effects of Glycyrrhiza
chalcones: experimental and computational studies. Phytomedicine.
18:795–801. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kwon HJ, Kim HH, Ryu YB, et al: In vitro
anti-rotavirus activity of polyphenol compounds isolated from the
roots of Glycyrrhiza uralensis. Bioorg Med Chem.
18:7668–7674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu TY, Khor TO, Saw CL, et al:
Anti-inflammatory/anti-oxidative stress activities and differential
regulation of Nrf2-mediated genes by non-polar fractions of tea
Chrysanthemum zawadskii and licorice Glycyrrhiza
uralensis. AAPS J. 13:1–13. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kao TC, Shyu MH and Yen GC:
Neuroprotective effects of glycyrrhizic acid and
18beta-glycyrrhetinic acid in PC12 cells via modulation of the
PI3K/Akt pathway. J Agric Food Chem. 57:754–761. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang W, Hu X, Zhao Z, et al:
Antidepressant-like effects of liquiritin and isoliquiritin from
Glycyrrhiza uralensis in the forced swimming test and tail
suspension test in mice. Prog Neuropsychopharmacol Biol Psychiatry.
32:1179–1184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen ZA, Wang JL, Liu RT, et al:
Liquiritin potentiate neurite outgrowth induced by nerve growth
factor in PC12 cells. Cytotechnology. 60:125–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun YX, Tang Y, Wu AL, et al:
Neuroprotective effect of liquiritin against focal cerebral
ischemia/reperfusion in mice via its antioxidant and antiapoptosis
properties. J Asian Nat Prod Res. 12:1051–1060. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Traynelis SF, Wollmuth LP, McBain CJ, et
al: Glutamate receptor ion channels: structure, regulation, and
function. Pharmacol Rev. 62:405–496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nicholls DG: Mitochondrial dysfunction and
glutamate excitotoxicity studied in primary neuronal cultures. Curr
Mol Med. 4:149–177. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jang JY, Kim HN, Kim YR, et al: Hexane
extract from Polygonum multiflorum attenuates
glutamate-induced apoptosis in primary cultured cortical neurons. J
Ethnopharmacol. 145:261–268. 2013.
|
|
12
|
Zhang M, Li J, Geng R, et al: The
inhibition of ERK activation mediates the protection of
necrostatin-1 on glutamate toxicity in HT-22 cells. Neurotox Res.
24:64–70. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin YL, Wang GJ, Huang CL, et al:
Ligusticum chuanxiong as a potential neuroprotectant for
preventing serum deprivation-induced apoptosis in rat
pheochromocytoma cells: functional roles of mitogen-activated
protein kinases. J Ethnopharmacol. 122:417–423. 2009. View Article : Google Scholar
|
|
15
|
Lou H, Fan P, Perez RG and Lou H:
Neuroprotective effects of linarin through activation of the
PI3K/Akt pathway in amyloid-β-induced neuronal cell death. Bioorg
Med Chem. 19:4021–4027. 2011.PubMed/NCBI
|
|
16
|
Lu S, Lu C, Han Q, et al: Adipose-derived
mesenchymal stem cells protect PC12 cells from glutamate
excitotoxicity-induced apoptosis by upregulation of XIAP through
PI3-K/Akt activation. Toxicology. 279:189–195. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin Y, Yan EZ, Fan Y, et al:
Neuroprotection by sodium ferulate against glutamate-induced
apoptosis is mediated by ERK and PI3 kinase pathways. Acta
Pharmacol Sin. 28:1881–1890. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mosmann T: Rapid colorimetric assay for
cellular growth and survival: application to proliferation and
cytotoxicity assays. J Immunol Methods. 65:55–63. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cossarizza A, Baccarani-Contri M,
Kalashnikova G and Franceschi C: A new method for the
cytofluorimetric analysis of mitochondrial membrane potential using
the J-aggregate forming lipophilic cation
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine
iodide (JC-1). Biochem Biophys Res Commun. 197:40–45.
1993.PubMed/NCBI
|
|
20
|
Yang CL, Chik SC, Li JC, Cheung BK and Lau
AS: Identification of the bioactive constituent and its mechanisms
of action in mediating the anti-inflammatory effects of black
cohosh and related Cimicifuga species on human primary blood
macrophages. J Med Chem. 52:6707–6715. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee CS, Kim YJ, Lee MS, Han ES and Lee SJ:
18beta-Glycyrrhetinic acid induces apoptotic cell death in SiHa
cells and exhibits a synergistic effect against antibiotic
anti-cancer drug toxicity. Life Sci. 83:481–489. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ricci JE, Gottlieb RA and Green DR:
Caspase-mediated loss of mitochondrial function and generation of
reactive oxygen species during apoptosis. J Cell Biol. 160:65–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dejean LM, Martinez-Caballero S and
Kinnally KW: Is MAC the knife that cuts cytochrome c from
mitochondria during apoptosis? Cell Death Differ. 13:1387–1395.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dudek H, Datta SR, Franke TF, et al:
Regulation of neuronal survival by the serine-threonine protein
kinase Akt. Science. 275:661–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dal-Cim T, Molz S, Egea J, et al:
Guanosine protects human neuroblastoma SH-SY5Y cells against
mitochondrial oxidative stress by inducing heme oxigenase-1 via
PI3K/Akt/GSK-3β pathway. Neurochem Int. 61:397–404. 2012.PubMed/NCBI
|
|
27
|
Ha T, Hua F, Liu X, et al:
Lipopolysaccharide-induced myocardial protection against
ischaemia/reperfusion injury is mediated through a
PI3K/Akt-dependent mechanism. Cardiovasc Res. 78:546–553. 2008.
View Article : Google Scholar
|
|
28
|
Ahmed NN, Grimes HL, Bellacosa A, Chan TO
and Tsichlis PN: Transduction of interleukin-2 antiapoptotic and
proliferative signals via Akt protein kinase. Proc Natl Acad Sci
USA. 94:3627–3632. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chao DT and Korsmeyer SJ: BCL-2 family:
regulators of cell death. Annu Rev Immunol. 16:395–419. 1998.
View Article : Google Scholar
|
|
30
|
Boucher MJ, Morisset J, Vachon PH, Reed
JC, Lainé J and Rivard N: MEK/ERK signaling pathway regulates the
expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of
human pancreatic cancer cells. J Cell Biochem. 79:355–369. 2000.
View Article : Google Scholar : PubMed/NCBI
|