Introduction
Colorectal cancer (CRC), which mainly originates
from the intestinal epithelium as premalignant lesions, termed
adenomas, is one of the most commonly diagnosed cancers in males
and females worldwide (1). Despite
prompt surgical removal followed by adjuvant therapy, which is
often suitable in the early stages of the disease, the majority of
patients still undergo therapeutic resistance and recurrence.
Therefore, investigation of the genetic and molecular mechanisms
that contribute to the progression and metastasis of CRC is
urgently required.
Previous evidence suggests that cancer progression
and metastasis are tightly associated with epithelial-mesenchymal
transition (EMT). EMT is a developmental process that involves
actin cytoskeleton reorganization and the loss of apical-basal
polarity and cell-to-cell contact, resulting in the conversion of
epithelial cells to mesenchymal cells (2,3).
Cells undergoing EMT are characterized by the loss of E-cadherin
and other components of epithelial cell junctions (4), and the acquisition of mesenchymal
markers, including N-cadherin and Vimentin (5). A series of EMT-inducing transcription
factors, including the zinc-finger proteins Snail and Slug, the
bHLH factor Twist, the zinc-finger/homeodomain proteins ZEB1 and
ZEB2, and the forkhead factor FoxC3, have been associated with
tumor invasion and metastasis (6).
Twist1, like other EMT-inducing transcription factors, including
Snail, Slug and SIP1, binds DNA through similar E-box sequence
motifs and represses E-cadherin and other epithelial cell adhesion
molecules. Further studies on different hepatic cellular cancer
(HCC) cell lines revealed that Twist1 is able to downregulate
E-cadherin expression and promote matrix metalloproteinase (MMP)
activation, specifically in MMP2 and MMP9 (7). Transforming growth factor (TGF)-β1 is
a multi-functional cytokine with diverse effects on cancer cells
(8). TGF-β1 is perhaps the most
common inducer of EMT (9,10). Although stimuli, including TGFβ,
FGF, EGF, IGF, HGF, PDGF, estrogens, Wnt, Shh, inflammatory
cytokines or hypoxia, as well as oncogenes like RasV12,
ErbB2 or mutant p53, may induce EMT during cancer progression
(3), the triggering and
maintenance of EMT is mainly orchestrated by three major groups of
transcription factors: the ZEB, Snail and Twist families (11).
Peroxiredoxins (PRDXs) constitute a family of
antioxidant enzymes that control cytokine-induced peroxide levels,
which mediate signal transduction in mammalian cells (12). As cancer cells are known to produce
large amounts of reactive oxygen species (ROS), it is readily
appreciated that overexpression of the antioxidant enzyme,
peroxiredoxins, may be beneficial for cancer cell survival. In
addition to the importance of eliminating oxidants, recent evidence
has begun to implicate other specific roles of antioxidant enzymes
in cellular functions, via protein-protein interaction or
controlling the local redox environment (13). However, the functions of
peroxiredoxins are not restricted to their antioxidant activities.
A number of studies have revealed novel functions in self-defense
against infection, tissue damages and tumors, through the
regulation of inflammation (14).
PRDX2, which is a typical 2-Cys thioredoxin
peroxidase and a cellular antioxidant that is widely distributed in
tissues, has been demonstrated to be overexpressed in various types
of cancer cells and tumor tissues. By virtue of its role in
maintaining the redox state in vivo, PRDX2 is a prime
candidate for regulating the H2O2 signaling
that is initiated by cell surface receptors (15). Previously, it has been well
demonstrated that PRDX2 may have a protective role against skin
aging and cellular senescence (16). Further characterization has
revealed that PRDX2 is present in the nucleus and enhances
agent-induced activation of the JNK/c-Jun pathway involved in
repair of damaged DNA (17). By
contrast, PRDX2 was recently demonstrated to function as a negative
regulator of PDGF signaling by suppressing the proliferation and
migration of smooth muscle cells (SMCs) (18). Another study also suggested that
PRDX2 acts as a growth suppressor to slow the induction of leukemia
caused by the c-Myc oncogene (19). These studies indicate that PRDX2
may be applicable as not only a predictive biomarker, but also a
potential therapeutic target.
However, to the best of our knowledge, evidence of a
possible correlation between PRDX2 expression and the EMT process
has not been published. The results from the present study
indicated that PRDX2 negatively regulates this process through a
pathway involving the transcription factors Twist1, Snail, ZEB1 and
ZEB2. Furthermore, the results demonstrated that upregulation of
PRDX2 may be correlated with EMT and contribute to the pathogenesis
of CRC by inhibiting EMT, cell migration and metastasis.
Materials and methods
Cell culture
The colorectal cancer cell lines, SW480 and SW620,
were respectively derived from tumor stage Dukes’ type B and C
human colorectal adenocarcinoma, which were both purchased from
American Type Culture Collection (ATCC, Manassas, VA, USA) and
maintained in Leibovitz’s 15 (L-15) cell culture medium (Gibco,
Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS;
SH30084.03; Hyclone, Logan, UT, USA) at 37°C in a humidified
atmosphere with 5% (v/v) CO2.
Reagents and antibodies
Recombinant human TGF-β1 (AF-100-21C) was purchased
from PeproTech (Rocky Hill, NJ, USA). The anti-hPRDX2 antibody
(EPR5154) was obtained from Abcam (Cambridge, MA, USA). Rabbit
monoclonal anti-human antibodies to E-cadherin (CDH1), N-cadherin
(CDH2), Vimentin (EPR3776), MMP2 (EPR1184) and MMP9 (EP1254) were
purchased from Epitomics (Abcam, San Francisco, CA, USA).
Anti-human antibodies against Twist1 (H-81), Snail1 (H-130) and
Slug (A-7) were obtained from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). Mouse monoclonal anti-human GAPDH and β-actin
antibodies were obtained from Tianjin Sungene Biotech, Co., Ltd.
(Tianjin, China). The HRP-conjugated goat anti-mouse or anti-rabbit
antibodies were purchased from Beyotime Institute of Biotechnology
(Shanghai, China). DyLightTM 488 AffiniPure goat
anti-rabbit secondary antibody and 4,6-diamidino-2-phenylindole
dihydrochloride (DAPI; blue fluorescence) were purchased from
EarthOx (San Francisco, CA, USA). The SYBR Green Quantitative PCR
kit was obtained from Takara (Dalian, China).
Cloning and transfection
The coding region of the human PRDX2 gene was cloned
from a human cDNA library (GenBank Accession no. NM_005809),
full-length PRDX2 cDNA was polymerase chain reaction
(PCR)-amplified using the following oligonucleotide primers:
PRDX2-F: 5′-GAGGATCCCCGGGTACCGGTCGCCACCATGGCCTCCGGTAACGC-3′
PRDX2-R: 5′-TCACCATGGTGGCGACCGGATTGTGTTTGGAGAAATATTCC-3′. Briefly,
the resulting fragment was ligated into the
pUbi-MCS-EGFP-IRES-Puromycin plasmid (Shanghai GeneChem, Shanghai,
China) to produce the lentiviral vector
pUbi-PRDX2-MCS-EGFP-IRES-Puromycin, which was named LV-PRDX2. As a
control, a lentiviral vector that expresses EGFP alone was also
generated and was named Vector or LV-CON. The final recombinant
lentiviral construct also was confirmed by DNA sequencing
analysis.
When the cells reached ~70–80% confluence, they were
transfected by Lipofectamine 2000 (Invitrogen Life Technologies,
Carlsbad, CA, USA) in accordance to the manufacturer’s
instructions. At 72 h post-transfection, the cells were observed
under a fluorescence microscope and the functional overexpression
of PRDX2 was confirmed by western blot analysis.
Cell migration assay
Cell migration assays were performed using a
two-chamber transwell device (Costar, Corning, USA) according to
the manufacturer’s instructions. After the cells were transfected
by LV-PRDX2 or LV-CON for 96 h, 50,000 cells that were suspended in
L-15 medium containing 0.1% FBS were seeded onto the upper chamber
of the device, and 500 μl of L-15 medium containing 10% FBS was
added to the lower chamber; the device was incubated at 37°C for 36
h. The inner side of the upper chamber was then wiped with a wet
cotton swab to remove the cells, while the outer side of the
chamber was gently rinsed with phosphate-buffered saline (PBS) and
treated with 4% paraformaldehyde for 20 min. The membrane was then
rinsed with PBS again and stained with a 0.1% crystal violet
staining solution for 30 min. After drying, >3 fields (between 3
and 8 fields) of the membrane were photographed, and the cells that
migrated through the membrane were counted in the lower wells of
the chamber by Countess® Automated Cell Counter
(Invitrogen Life Technologies). Each experiment was performed three
times.
RNA isolation and quantitative polymerase
chain reaction (qPCR)
Total RNA was isolated using TRIzol reagent
(Invitrogen Life Technologies). Total RNA concentration was
determined spectrophotometrically at 260 and 280 nm. cDNA was
synthesized using the TaqMan miRNA Reverse Transcription kit
(Qiagen, Valencia, CA, USA). The mRNA level was determined using
the SYBR PrimeScript RT-PCR kit (Takara).
qPCR was performed using the QPCR SYBR Green Low ROX
Mix (Thermo Fisher Scientific Inc., Rockford, IL, USA) according to
the manufacturer’s instructions. The mRNA expression levels of the
target genes and GAPDH as a normalizing control were analyzed using
the following primer sets: E-cadherin, forward:
5′-CTTCTCTCACGCTGTGTCATC-3′ and reverse:
5′-CTCCTGTGTTCCTGTTAATGGT-3′; N-cadherin, forward: 5′-CGAATGGATG
AAAGACCCATCC-3′ and reverse: 5′-GGAGCCACTGCC TTCATAGTCAA-3′;
Vimentin, forward: 5′-AGGCAAAGCAGGAGTCCA-3′ and reverse:
5′-TATCAACCAGAGGGAGTG-3′; Twist1, forward:
5′-GGCTCAGCTACGCCTTCTC-3′ and reverse:
5′-TCCTTCTCTGGAAACAATGACA-3′; Snail, forward:
5′-GACCACTATGCCGCGCTCTT-3′ and reverse:
5′-TCGCTGTAGTTAGGCTTCCGATT-3′; Slug, forward:
5′-TGGTTGCTTCAAGGACACAT-3′ and reverse: 5′-GCAAATGCTCTGTTGCAGTG-3′;
ZEB1, forward: 5′-TACAGAACCCAACTTGAACGTCACA-3′ and reverse
5′-GATTACACCCAGACTGCGTCACA-3′; ZEB2, forward:
5′-CACAGCTCTTCCACCTCAAAGC-3′ and reverse:
5′-TTTTGCGAGACAGACAGGAG-3′ and GAPDH, forward:
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse: 5′-TGGTGAAGACGCCAGTGGA-3′.
Thermal cycling conditions comprised 1 step for 10 min at 95°C
followed by 40 cycles for 15 sec at 95°C and for 1 min at 60°C.
Each measurement was performed in duplicates and the PCR product
quality was monitored through post-PCR melt-curve analysis. The
relative values of gene expression were normalized to that of GAPDH
and calculated using the 2−ΔΔCt method, where ΔΔCt =
(ΔCt target gene − ΔCtGAPDH)sample
− (ΔCttarget gene −
ΔCtGAPDH)control. The fold change in relative
expression was then determined by calculating
2−ΔΔCt.
Western blot analysis
Total proteins were isolated from frozen tissues
using lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 10 mM EDTA, 1%
NP-40, 0.1% SDS, 1 mM PMSF and 0.5% sodium deoxycholate), and
quantified using the Bradford method. The cell lysates were
separated on 8 or 12% SDS-PAGE gel and transferred onto a PVDF
membrane at 90 V for 150 min. The blots were incubated with
anti-E-cadherin, anti-N-cadherin, anti-Vimentin, anti-Twist1,
anti-Snail, anti-Slug, anti-MMP2 and anti-MMP9 antibodies and
developed using the enhanced chemiluminescence (ECL) detection
system (Bio-Rad, Hercules, CA, USA). The same blot was stripped and
reprobed with the anti-β-actin antibody for use as an internal
control.
Statistical analysis
Data are presented as the mean ± standard deviation
of three independent experiments, and the SPSS version 18 software
package was used for statistical analysis (SPSS, Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Forced PRDX2 expression blocks
TGF-β1-induced EMT and cell migration
To investigate whether PRDX2 is involved in
TGF-β1-induced EMT, its expression was transiently forced in CRC
cells using lentivirus vectors. LV-PRDX2 or LV-CON were transfected
into colorectal cancer SW480 cells, then the morphological changes
were observed with phase-contrast microscopy and the expression of
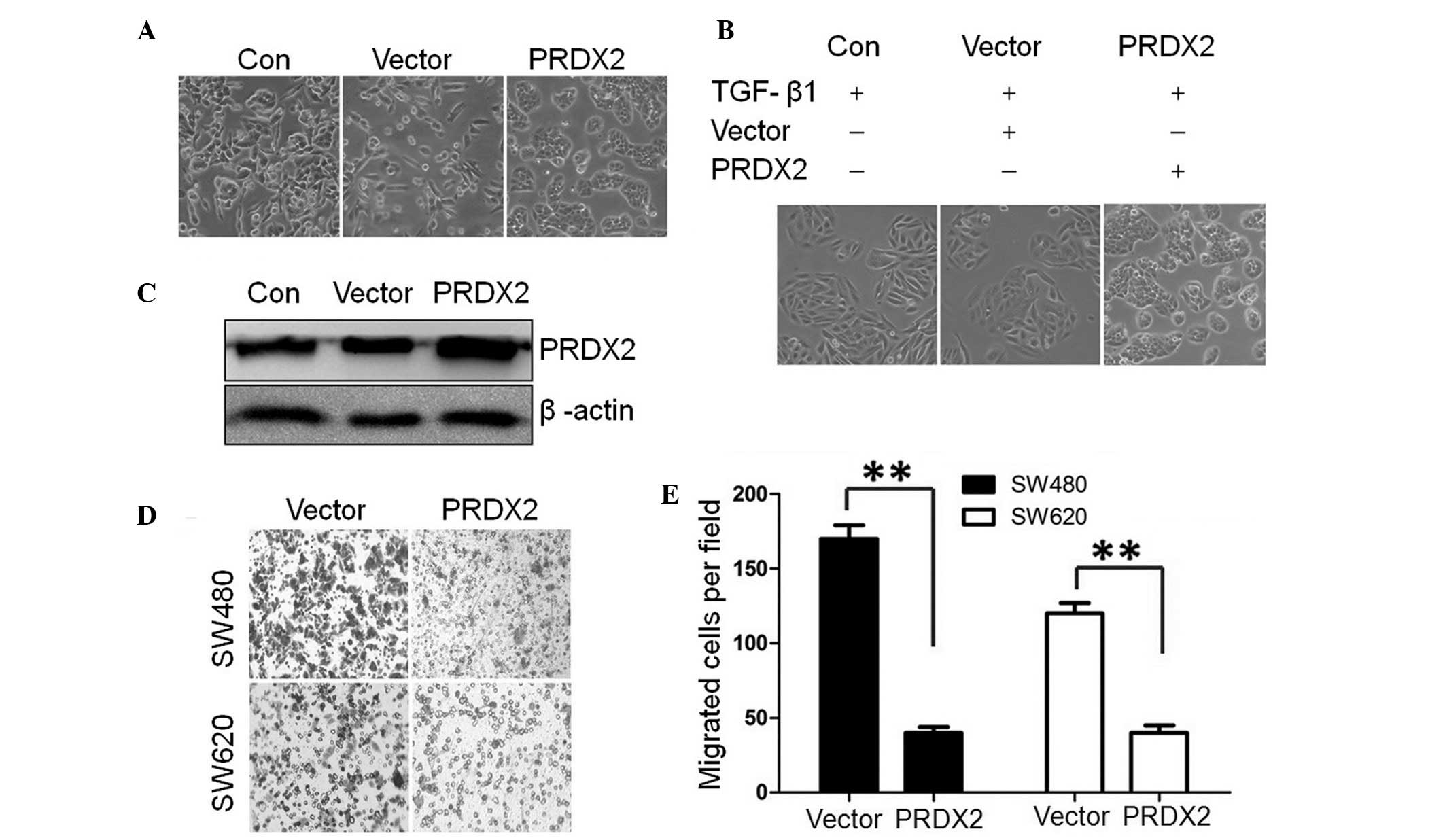
PRDX2 was detected by western blotting (Fig. 1A and C). Compared with the
untreated cells (Con) and lentiviral vector control (Vector), the
SW480 cells transfected by LV-PRDX2 exhibited a change of cell
accumulation. As observed, the SW480 cells treated with TGF-β1 for
72 h lead to a morphological change from an epithelial phenotype to
an elongated, spindle-like mesenchymal phenotype. These findings
demonstrated that TGF-β1 may induce EMT-like morphological changes
in SW480 cells. Compared with the control group, the spindle-like
morphological changes were not observed upon TGF-β1 addition in
LV-PRDX2 transfected cells (Fig.
1B). The result demonstrated that PRDX2 overexpression
eliminated the EMT-like morphological changes of the SW480
cells.
To determine the role of PRDX2 during the EMT-like
process, cell migration assays were performed (Fig. 1D). Further analysis demonstrated
that forced PRDX2 expression markedly reduced the TGF-β1-increased
cell migration in the two cell lines compared with the control
group (P<0.01; Fig. 1E).
Collectively, these results demonstrated that forced PRDX2 may
inhibit TGF-β1-induced EMT-like phenotype and cell migration in
colorectal cancer cells.
PRDX2 regulates the expression of EMT
markers and EMT-related transcription factors in colorectal cancer
cells at the mRNA level
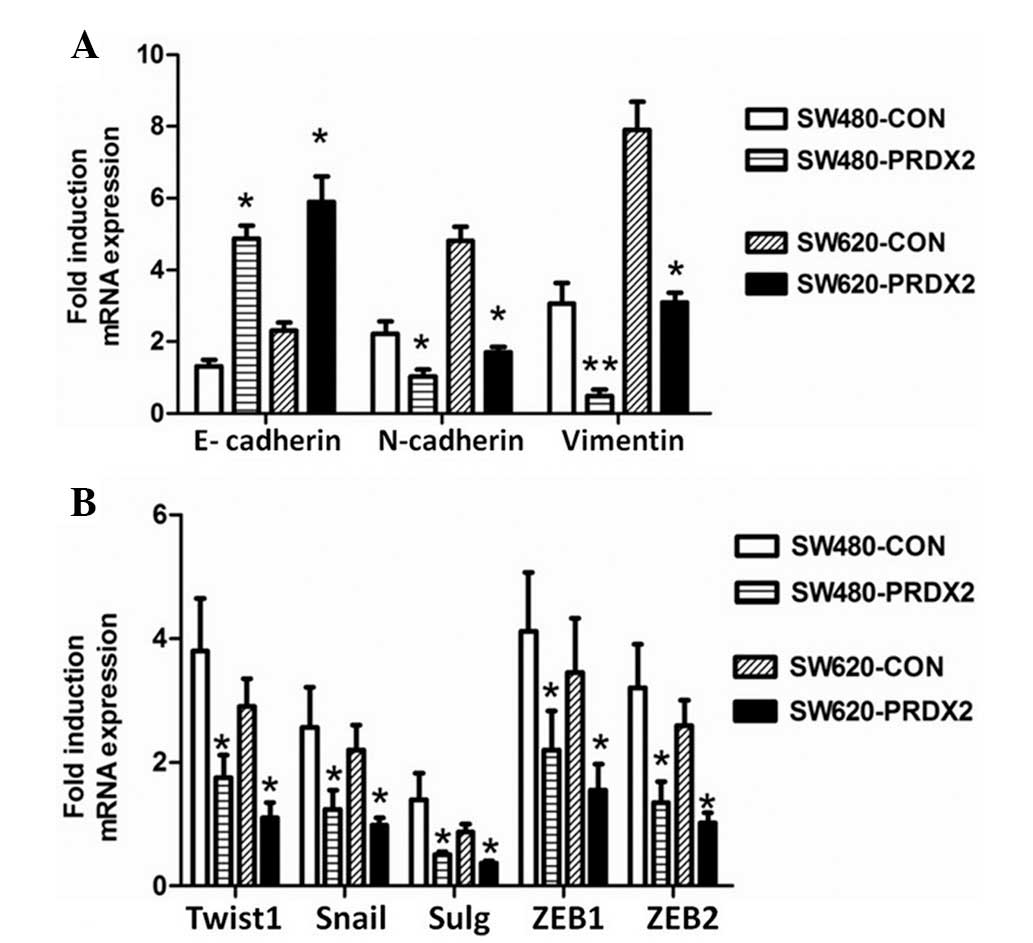
To determine the function of PRDX2 during the
EMT-like process in vitro, qPCR analysis for EMT markers and
EMT-related transcription factors was performed. As demonstrated in
Fig. 2A, the mRNA level of
E-cadherin was markedly higher compared with the control group in
the SW480 and SW620 cells. However, the mRNA level of the
mesenchymal markers, N-cadherin and Vimentin, were significantly
reduced in comparison with those in the control cells. Compared
with the control group, overexpression of PRDX2 significantly
increased the mRNA level of the EMT-related transcription factors
Twist1, Snail, Slug, ZEB1 and ZEB2 in CRC cells stimulated with
TGF-β1 for 72 h (Fig. 2B). These
results revealed that PRDX2 regulates EMT markers and EMT-related
transcription factors in CRC cells at the mRNA level.
Overexpression of PRDX2 modulates the
expression of TGF-β1-induced EMT markers and EMT-related
transcription factors and metastasis-related factors
To investigate the molecular mechanism of how the
overexpression of PRDX2 modulates the expression of TGF-β1-induced
EMT, the expression of EMT marker, EMT master transcription factors
and metastasis-related factors was examined using western blot
analysis. As predicted, the overexpression of PRDX2 resulted in the
upregulation of the E-cadherin and downregulation of the N-cadherin
and Vimentin. Furthermore, its overexpression inhibited the
expression of the transcription factors Twist1, Snail, Slug and
metastasis-related factors MMP2 and MMP9 in CVC cells stimulated
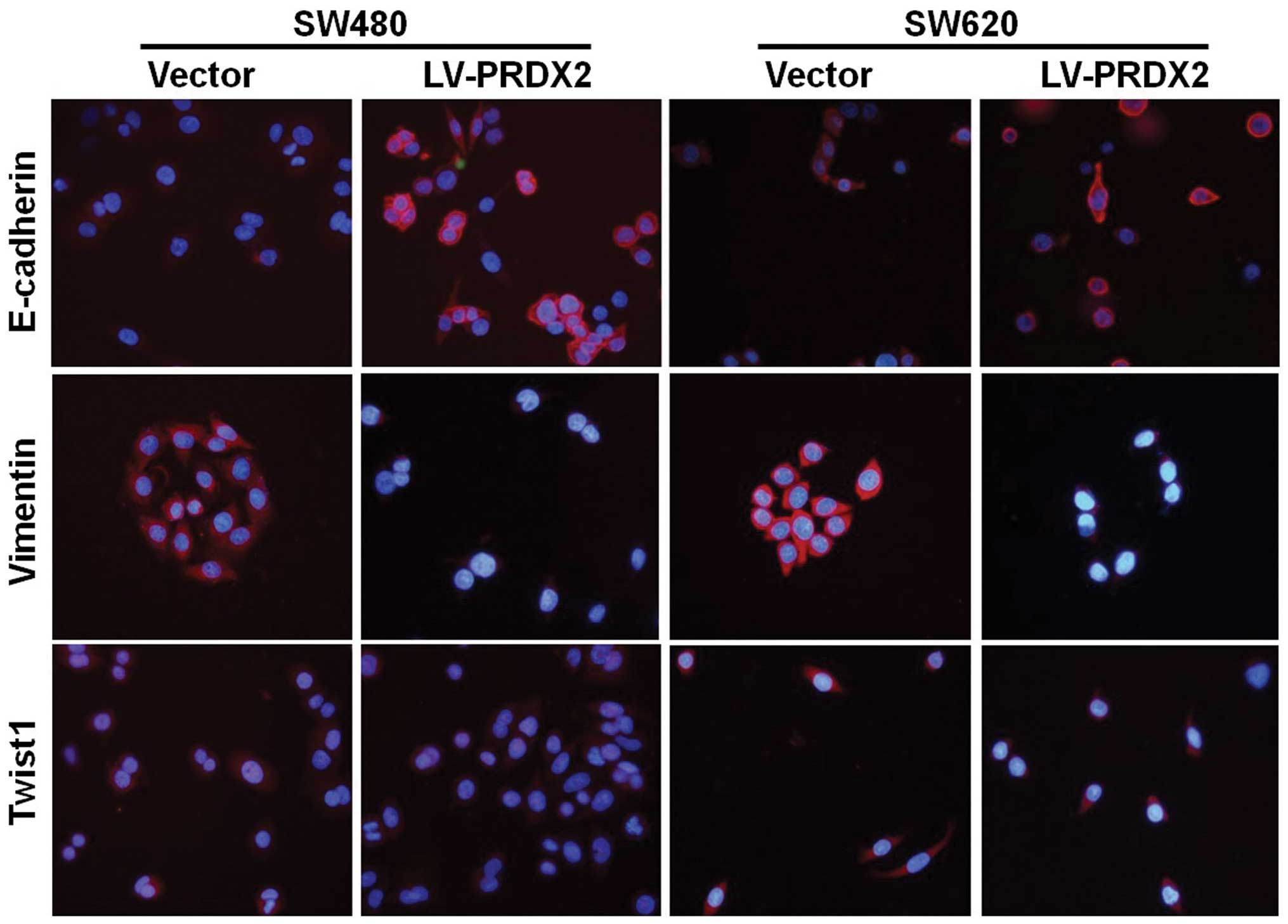
with TGF-β1 (Fig. 3). Next, the
regulatory effects of PRDX2 on EMT markers and the transcription
factors Twist1 were further confirmed using immunofluorescence
staining. The results suggested that PRDX2 may have an important
role in the EMT-like process by upregulating E-cadherin and
downregulating Vimentin and Twist1 (Fig. 4). Together, these data further
indicated that overexpression of PRDX2 modulates TGF-β1-induced EMT
in CRC cells.
 | Figure 3Expression of E-cadherin, N-cadherin,
Vimentin, Twist1, Snail, Slug, MMP2 and MMP9 were analyzed by
western blotting, and β-actin was used as a loading control. NC,
negative control; LV-Con, lentiviral vector control; PRDX2,
peroxiredoxin 2; MMP, matrix metalloproteinase. |
Discussion
In the past decade, numerous studies have reported
that the EMT process is a morphological event that is crucial to
tumor migration and invasion in physiological and pathological
states (20,21). TGF-β1 has an important role in
tumor progression by inducting EMT (4,22).
Although it is well established that PRDX2 is involved in cancer
progression, little is known regarding the function of PRDX2 in the
EMT-like process of tumor cells.
In the present study, it was identified that
overexpression of PRDX2 inhibits CRC cell invasion and
TGF-β1-induced EMT. Therefore, the upregulation of PRDX2 may cause
MET in CRC cells, which is the reverse process of EMT, and
associated with cell migration and metastasis. Although the cell
morphological changes associated with EMT in PRDX2 knockdown cells
were not studied, the present data suggested that forced PRDX2
expression modulates the expression of TGF-β1-induced EMT markers
and its related transcription factors, which indicates a potential
role of PRDX2 in TGF-β1-mediated biological functions. In addition,
PRDX2 negatively regulated the metastasis-related factors, MMP2 and
MMP9, suggesting that PRDX2 may be a new regulator of metastasis in
CRC patients.
Previously, the expression of peroxiredoxins in
mammalian cells, particularly peroxiredoxin1 (PRDX1) and PRDX2, has
been considered to be indicative of signal peroxidases receiving,
transducing and transmitting peroxide signals (23–25).
However, a similar response of the ASK1-p38 pathway in knocking
down PRDX1 and overexpressing PRDX2 suggested that the two
cytosolic peroxiredoxins have distinct roles in the cellular
peroxide response (23).
Previously, one study reported that PRDX1 overexpression may
enhance the TGF-β1-induced EMT and cell migration, whereas
knockdown of PRDX1 significantly inhibited the TGF-β1-induced EMT
and cell migration in lung cancer cells (26). These findings proved that PRDX2 and
PRDX1 may have opposing roles in the regulation of the
TGF-β1-induced EMT and cell migration in cancer cells. Further
investigations are required to determine the molecular link between
the two 2-Cys peroxiredoxins and EMT-related transcription
factors.
It has recently been demonstrated that the
progression from normal intestinal mucosa to adenoma (adenomatous
mucosa) and finally to adenocarcinoma in CRC is closely correlated
with the EMT process and changes in the expression of a series of
genes, including E-cadherin, Vimentin and β-catenin (27). Another study revealed that PRDX2
may be a novel invasion and metastasis suppressor due to its
ability to increase the formation of E-cadherin/β-catenin complexes
in melanoma (28). Our previous
studies demonstrated that overexpression of PRDX2 promoted cell
proliferation and prevented oxidation-induced apoptosis (29), whereas silencing of PRDX2
expression led to increased apoptosis and decreased proliferation
by the Wnt/β-catenin pathway in CRC cells (30). Furthermore, upregulation of PRDX2
resulted in the elimination of intracellular
H2O2 (31),
which negatively modulated the Wnt signal pathway by downregulating
β-catenin (32). Key targets of
the pathways that induce EMT include the adherens junction
components E-cadherin and β-catenin. Therefore, PRDX2 may be
involved in cell adhesion by stabilizing the E-cadherin/β-catenin
complexes, and overexpression of PRDX2 may repress EMT and cell
migration by activating the Wnt/β-catenin pathways. This hypothesis
is consistent with the behavior of PRDX2 in modulating the EMT
signal pathway. In vivo and in vitro model systems
have allowed the characterization of various pathways leading to
EMT and EMT-like phenotypes. Downregulation of E-cadherin is a
critical initial step in EMT, not only because of the disruption of
adherens junctions but also because loss of E-cadherin reinforces
the EMT process by inducing the expression of Twist1 and ZEB1 in a
feed-forward loop (33). As
EMT-activating transcription factors, particularly Snail, Slug,
Twist, SIP1/ZEB are able to negatively regulate the expression of
E-cadherin (34). Although the
transcriptional regulatory networks that orchestrate the EMT
process remain unclear, it is possible that PRDX2 may regulate the
signaling pathways that induce E-cadherin regulators and modulate
the EMT-related transcriptional factors. Extensive functional
studies are therefore required to select candidate proteins for
further validation.
In conclusion, although the mechanism of PRDX2 that
is involved in the EMT-like process has not been fully illustrated,
the data implicated PRDX2 in transcriptional regulation of the
TGF-β1-induced EMT and cell migration. The present findings
demonstrated that PRDX2 may have a crucial role by impeding EMT in
CRC cells, and implies that it represses cell migration and
metastasis of CRC cells by controlling the expression of genes that
are crucial for the TGFβ1-induced EMT. In addition, when the
specific agents involved in the process of metastasis by inducing
EMT are further identified, therapeutic strategies may be developed
that target silencing of oncogenes or upregulating the EMT-mediated
gene. Designing therapeutics targeting PRDX2 may offer a novel
strategy for developing treatments for improving the prognosis for
highly malignant types of CRC.
Acknowledgements
This study was supported by a grant from the Natural
Science Foundation of China (grant no. 81172295). The authors are
grateful to the editors of AJE for kindly revising our paper.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Developmental Cell. 14:818–829. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Massagué J: TGFbeta in Cancer. Cell.
134:215–230. 2008.
|
|
5
|
Stefan Grünert MJHB: Diverse cellular and
molecular mechanisms contribute to epithelial plasticity and
metastasis. Nat Rev Mol Cell Biol. 4:657–665. 2003.PubMed/NCBI
|
|
6
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.PubMed/NCBI
|
|
7
|
Zhao XL, Sun T, Che N, et al: Promotion of
hepatocellular carcinoma metastasis through matrix
metalloproteinase activation by epithelial-mesenchymal transition
regulator Twist1. J Cell Mol Med. 15:691–700. 2011. View Article : Google Scholar
|
|
8
|
Cho KH, Jeong KJ, Shin SC, Kang J, Park CG
and Lee HY: STAT3 mediates TGF-beta1-induced TWIST1 expression and
prostate cancer invasion. Cancer Lett. 336:167–173. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kalluri RWR: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Akhurst RJ and Derynck R: TGF-beta
signaling in cancer - a double-edged sword. Trends Cell Biol.
11:S44–S51. 2001.PubMed/NCBI
|
|
11
|
Sánchez-Tilló E, Liu Y, de Barrios O, et
al: EMT-activating transcription factors in cancer: beyond EMT and
tumor invasiveness. Cellular and molecular life sciences. Cell Mol
Life Sci. 69:3429–3456. 2012.PubMed/NCBI
|
|
12
|
Wood ZASE, Schröder E, Robin Harris J and
Poole LB: Structure, mechanism and regulation of peroxiredoxins.
Trends Biochem Sci. 28:32–40. 2003. View Article : Google Scholar
|
|
13
|
Kang DH, Lee DJ, Lee KW, et al:
Peroxiredoxin II is an essential antioxidant enzyme that prevents
the oxidative inactivation of VEGF receptor-2 in vascular
endothelial cells. Mol Cell. 44:545–558. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ishii T, Warabi E and Yanagawa T: Novel
roles of peroxiredoxins in inflammation, cancer and innate
immunity. J Clin Biochem Nutr. 50:91–105. 2012.PubMed/NCBI
|
|
15
|
Rhee SG, Chae HZ and Kim K:
Peroxiredoxins: a historical overview and speculative preview of
novel mechanisms and emerging concepts in cell signaling. Free
Radic Biol Med. 38:1543–1552. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han YH, Kim HS, Kim JM, Kim SK, Yu DY and
Moon EY: Inhibitory role of peroxiredoxin II (Prx II) on cellular
senescence. FEBS Letters. 579:4897–4902. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee KW, Lee DJ, Lee JY, Kang DH, Kwon J
and Kang SW: Peroxiredoxin II restrains DNA damage-induced death in
cancer cells by positively regulating JNK-dependent DNA repair. J
Biol Chem. 286:8394–8404. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi MH, Lee IK, Kim GW, et al: Regulation
of PDGF signalling and vascular remodelling by peroxiredoxin II.
Nature. 435:347–353. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Agrawal-Singh S, Isken F, Agelopoulos K,
et al: Genome-wide analysis of histone H3 acetylation patterns in
AML identifies PRDX2 as an epigenetically silenced tumor suppressor
gene. Blood. 119:2346–2357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tiwari N, Gheldof A, Tatari M and
Christofori G: EMT as the ultimate survival mechanism of cancer
cells. Semin Cancer Biol. 22:194–207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y and Shang Y: Epigenetic control of
epithelial-to-mesenchymal transition and cancer metastasis. Exp
Cell Res. 319:160–169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ko H, So Y, Jeon H, et al:
TGF-beta1-induced epithelial-mesenchymal transition and acetylation
of Smad2 and Smad3 are negatively regulated by EGCG in human A549
lung cancer cells. Cancer Lett. 335:205–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jarvis RM, Hughes SM and Ledgerwood EC:
Peroxiredoxin 1 functions as a signal peroxidase to receive,
transduce, and transmit peroxide signals in mammalian cells. Free
Radic Biol Med. 53:1522–1530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Manandhar G, Miranda-Vizuete A, Pedrajas
JR, et al: Peroxiredoxin 2 and peroxidase enzymatic activity of
mammalian spermatozoa. Biol Reprod. 80:1168–1177. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Neumann CA, Cao J and Manevich Y:
Peroxiredoxin 1 and its role in cell signaling. Cell Cycle.
8:4072–4078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ha B, Kim EK, Kim JH, et al: Human
peroxiredoxin 1 modulates TGF-β1-induced epithelial-mesenchymal
transition through its peroxidase activity. Biochem Biophys Res
Commun. 421:33–37. 2012.PubMed/NCBI
|
|
27
|
Chen X, Halberg RB, Burch RP and Dove WF:
Intestinal adenomagenesis involves core molecular signatures of the
epithelial-mesenchymal transition. J Mol Histol. 39:283–294. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee DJ, Kang DH, Choi M, et al:
Peroxiredoxin-2 represses melanoma metastasis by increasing
E-Cadherin/beta-Catenin complexes in adherens junctions. Cancer
Res. 73:4744–4757. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu W, Fu Z, Wang H, Feng J, Wei J and Guo
J: Peroxiredoxin 2 is upregulated in colorectal cancer and
contributes to colorectal cancer cells’ survival by protecting
cells from oxidative stress. Mol Cell Biochem. 387:261–270.
2014.PubMed/NCBI
|
|
30
|
Lu W, Fu Z, Wang H, Feng J, Wei J and Guo
J: Peroxiredoxin 2 knockdown by RNA interference inhibits the
growth of colorectal cancer cells by downregulating Wnt/β-catenin
signaling. Cancer Lett. 343:190–199. 2014.PubMed/NCBI
|
|
31
|
Johnson RM, Ho YS, Yu DY, et al: The
effects of disruption of genes for peroxiredoxin-2, glutathione
peroxidase-1, and catalase on erythrocyte oxidative metabolism.
Free Radic Biol Med. 48:519–525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shin SY, Kim CG, Jho EH, et al: Hydrogen
peroxide negatively modulates Wnt signaling through downregulation
of beta-catenin. Cancer Lett. 212:225–231. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Onder TT, Gupta PB, Mani SA, et al: Loss
of E-cadherin promotes metastasis via multiple downstream
transcriptional pathways. Cancer Res. 68:3645–3654. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu QC, Gao RY, Wu W and Qin HL:
Epithelial-mesenchymal transition and its role in the pathogenesis
of colorectal cancer. Asian Pac J Cancer Prev. 14:2689–2698. 2013.
View Article : Google Scholar : PubMed/NCBI
|